Rational Design of a Covalent EGFR T790M Inhibitor Using LiteFold



A structure-guided, AI-assisted computational workflow for covalent inhibitor design against a drug-resistant NSCLC mutation
The Clinical Imperative: The Evolutionary Arms Race in Non-Small Cell Lung Cancer
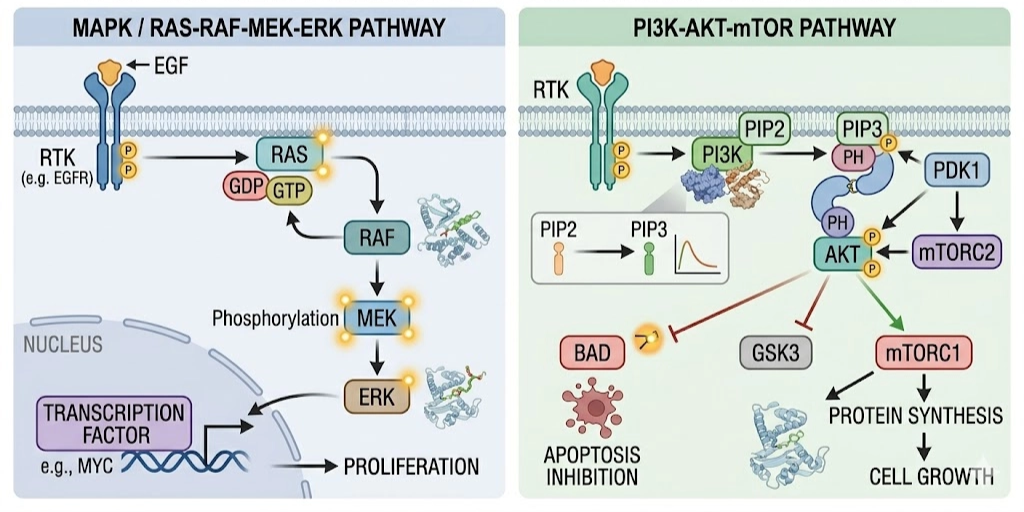
The rise of precision oncology has been driven by the understanding that specific genetic mutations can directly control cancer growth and survival. Targeted cancer therapy has greatly improved outcomes in non-small cell lung cancer (NSCLC), especially through inhibition of the Epidermal Growth Factor Receptor (EGFR). Activating mutations in EGFR, such as exon 19 deletions and L858R, lead to continuous signaling through growth pathways like RAS–RAF–MEK–ERK and PI3K–AKT, resulting in uncontrolled cell proliferation. However, in many lung adenocarcinomas, activating mutations in the EGFR kinase domain disrupt this regulation. The most common mutations, including exon 19 deletions and the L858R substitution, lock the receptor in a constitutively active state. As a result, cells receive continuous growth signals independent of external stimuli, leading to uncontrolled proliferation and tumor progression.
First-generation inhibitors such as Gefitinib and Erlotinib initially show strong clinical response by reversibly binding to the ATP-binding pocket. However, resistance develops in most patients within a year. The primary cause of this resistance is the T790M mutation, where threonine is replaced by methionine at position 790. This mutation increases ATP affinity and introduces a bulkier hydrophobic side chain, making it difficult for reversible inhibitors to bind effectively.
Third-generation inhibitors like Osimertinib address this issue by forming a covalent bond with Cys797. However, design challenges and resistance mechanisms still remain.
In this study, we aim to address these challenges through a structure-guided, AI-assisted design approach. Using the LiteFold platform, we perform de novo generation and optimization of small molecules tailored specifically to the EGFR T790M binding pocket. By integrating molecular docking, covalent design principles, and molecular dynamics simulations, we evaluate not only the binding affinity of the designed molecules but also their dynamic stability within the receptor environment. This approach allows us to move beyond static predictions and establish whether the designed inhibitor can maintain a stable pre-covalent state, a critical requirement for effective covalent inhibition.
First-generation reversible inhibitors Gefitinib and Erlotinib produce strong initial clinical responses, but acquired resistance develops in the majority of patients within approximately twelve months. The dominant resistance mechanism is the T790M gatekeeper mutation, in which threonine at position 790 is replaced by the bulkier methionine. This substitution simultaneously increases ATP-binding affinity and sterically hinders accommodation of reversible inhibitors, effectively restoring kinase activity despite drug treatment.
Third-generation inhibitors, most notably Osimertinib, circumvent this resistance by forming an irreversible covalent bond with Cysteine 797 (Cys797) in the ATP-binding pocket, and by doing so are selective for the T790M mutant over wild-type EGFR. Yet even this class is not immune to further resistance most notably through the C797S mutation, which ablates the nucleophilic cysteine entirely.
This study describes a structure-guided, AI-assisted workflow for de novo design and dynamic validation of a new covalent inhibitor candidate specifically tailored to the EGFR T790M binding pocket, using the LiteFold computational platform.
The Rationale for Covalent Inhibition
The pharmaceutical industry was historically cautious about intentionally reactive (electrophilic) molecules, owing to concerns over idiosyncratic toxicity, haptenization, and off-target covalent modification of unintended cellular proteins or nucleic acids. For decades, the prevailing view held that the risks of covalent targeting outweighed any benefits, a position complicated by the fact that several landmark drugs, including aspirin (cyclooxygenase inhibition) and the penicillin class (bacterial transpeptidase inhibition), owe their efficacy to serendipitous covalent mechanisms.
Advances in structural bioinformatics, chemoproteomics, and structure-guided design have substantially rehabilitated the field. The modern approach to targeted covalent inhibition (TCI) employs two integrated molecular features: a selective non-covalent recognition scaffold that precisely positions the molecule within the target site, and a mildly electrophilic warhead that reacts irreversibly only once the correct geometry is achieved.
In the context of EGFR T790M, the target nucleophile is Cys797, a non-catalytic residue at the solvent-exposed edge of the ATP-binding cleft. This residue is relatively rare across the broader kinome, making it an attractive orthogonal handle for achieving both potency and selectivity. Second-generation pan-HER inhibitors (Afatinib, Dacomitinib) and the mutant-selective third-generation inhibitor Osimertinib all exploit an acrylamide warhead to alkylate Cys797, extending progression-free survival in T790M-positive patients.
Inhibitor Generation Overview
| Class | Binding | Selectivity | Limitation |
|---|---|---|---|
| 1st gen (Erlotinib) | Reversible | WT + mutants | T790M resistance |
| 2nd gen (Afatinib) | Covalent (pan-HER) | WT + mutants | WT toxicity |
| 3rd gen (Osimertinib) | Covalent | T790M mutant | C797S resistance |
Computational Design Workflow
To address EGFR T790M resistance, a three-phase computational workflow was conducted using the LiteFold platform:
1. De novo molecular design — pocket-guided generation of candidate molecules
2. Molecular docking — evaluation of binding affinity and interaction geometry
3. Molecular dynamics (MD) simulation — assessment of dynamic stability in an explicit solvent environment
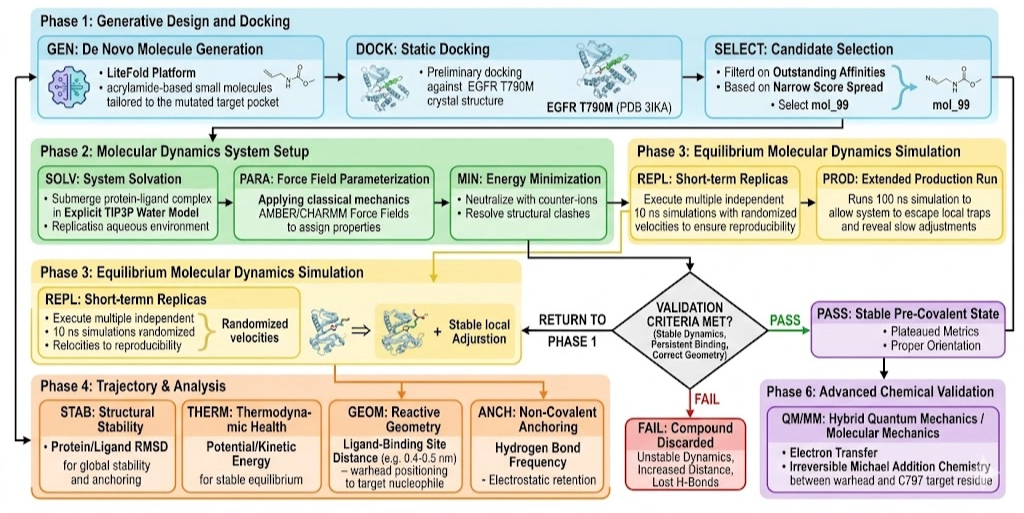
Phase 1: De Novo Design
LiteFold's de novo design module generates small molecules directly from the geometry and chemical character of a specified binding pocket, rather than by decorating known scaffolds. The EGFR T790M pocket (PDB ID: 3IKA) was used as the design template, with explicit attention to four key residues:
• Met790 — the gatekeeper mutation site, which introduces increased hydrophobicity and reduced pocket volume
• Met793 — the hinge region, a critical hydrogen-bond donor/acceptor anchor for kinase inhibitors
• Lys745 and Glu762 — catalytic residues that define the electrostatic environment of the ATP pocket
• Cys797 — the intended covalent target, positioned at the solvent-exposed lip of the binding cleft
The platform generated a library of candidate molecules that were immediately subjected to preliminary docking against the EGFR T790M crystal structure. The top five ranked compounds are shown below.
| Rank | Molecule | Score | Assessment | Selected |
|---|---|---|---|---|
| 1 | mol_88 | −9.71 | Outstanding | — |
| 2 | mol_65 | −8.52 | Excellent | — |
| 3 | mol_43 | −8.43 | Excellent | — |
| 4 | mol_85 | −8.20 | Very strong | — |
| 5 | mol_99 | −8.18 | Very strong | ✓ Selected |
Docking scores below −8.0 kcal/mol are generally associated with nanomolar-range binding affinities, though docking scores alone are not reliable quantitative predictors of experimental Ki values and should be interpreted with appropriate caution.
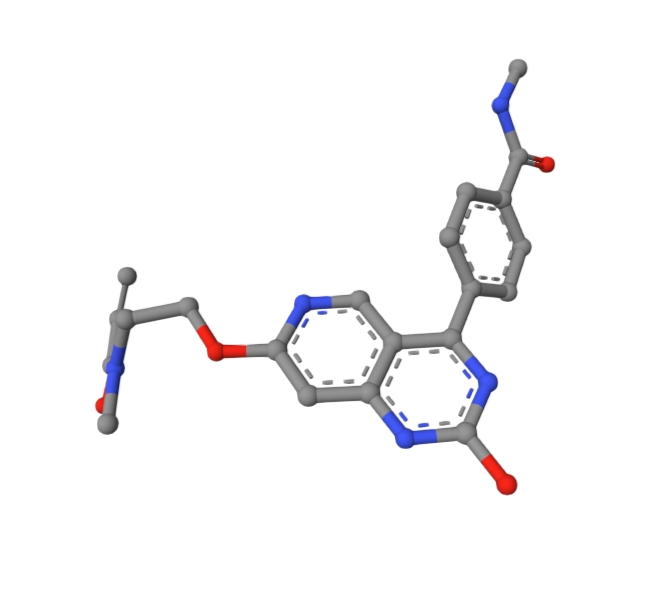
Despite not achieving the top docking score, mol_99 was selected as the primary scaffold for further development for three reasons: its interaction geometry placed the reactive vector of the nascent warhead in close proximity to the Cys797 sulfur; the scaffold demonstrated structural compatibility with acrylamide incorporation without predicted steric penalties; and its docking score spread across poses was narrow, suggesting a well-defined and reproducible binding mode rather than pose ambiguity.
Phase 2: Molecular Docking and Binding Evaluation
The mol_99 scaffold, modified to carry an acrylamide warhead at the appropriate exit vector (mol99_OV4_lead_covalent.sdf), was docked against the active conformation of EGFR T790M (PDB ID: 3IKA) to generate a detailed binding profile.
Across ten independently generated docking poses, the binding scores ranged from −6.84 to −6.35 kcal/mol a spread of approximately 0.5 kcal/mol. In computational drug design, a tight energetic cluster of this magnitude is a meaningful signal: it indicates that the molecule consistently samples a single binding mode within the ATP pocket rather than adopting multiple, potentially artifactual orientations. This pose consistency implies that the shape and electronic properties of the molecule are well-matched to the receptor's topography a hallmark of a robust lead candidate suitable for progression to dynamic simulation.
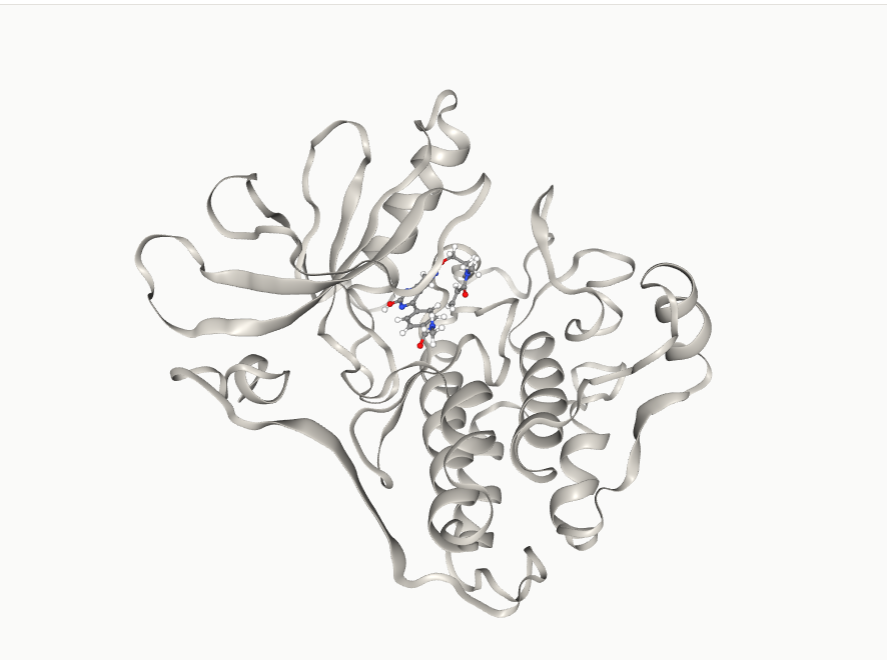
The Two-Step Kinetics of Covalent Inhibition
A critical conceptual point for interpreting both the docking and MD results is the two-step kinetic mechanism that governs all targeted covalent inhibitors.
Step 1 — Non-Covalent Association
The inhibitor must first enter the binding pocket and form a stable, reversible non-covalent complex (E·I). This step is governed entirely by classical intermolecular forces hydrogen bonding, van der Waals contacts, and hydrophobic packing and is described by the equilibrium dissociation constant Ki. Without a thermodynamically stable pre-covalent complex, the warhead is never positioned close enough to the target nucleophile for chemistry to occur.
Step 2 — Covalent Bond Formation
Only once the pre-covalent complex is stabilized can the electrophilic acrylamide warhead undergo Michael addition with the Cys797 thiolate, forming the irreversible covalent adduct (E–I). The rate of this irreversible step is governed by kinact, the maximal rate of enzyme inactivation. The overall potency of a targeted covalent inhibitor is therefore determined by the kinact/Ki ratio not by the warhead reactivity alone.
This two-step paradigm explains why static docking alone is insufficient to validate a covalent inhibitor candidate. A molecule may appear optimally positioned in a frozen crystal-structure pose, yet rapidly drift or dissociate when exposed to the thermodynamic realities of an aqueous environment: protein conformational flexibility, entropic penalties, and solvent competition. Establishing that the designed inhibitor maintains a stable pre-covalent orientation over biologically relevant timescales is therefore a prerequisite before proceeding to synthesis or advanced covalent modelling.
Phase 3: Molecular Dynamics Validation
Simulation Setup
Molecular dynamics (MD) simulation treats each atom as a classical particle obeying Newtonian mechanics. By computing interatomic forces and integrating Newton's equations of motion across femtosecond time steps, MD generates a continuous, high-resolution trajectory of atomic behaviour effectively serving as a computational microscope capable of resolving protein ligand dynamics that static docking cannot capture.
The solvation model is a particularly important methodological choice. Protein structure and function are inextricably coupled to the solvent environment: water molecules mediate hydrogen bonding networks, drive the hydrophobic effect that contributes to ligand binding, and determine the desolvation penalty that the Cys797 thiolate must pay before covalent attack. For this study, the system was solvated using the explicit TIP3P (Transferable Intermolecular Potential with 3 Points) water model a three-site rigid representation with partial charges on oxygen and hydrogen, combined with a Lennard–Jones potential centered on the oxygen. Although four- and five-site water models offer improved reproduction of certain bulk-water properties, TIP3P remains the standard for atomistic protein–ligand simulations in conjunction with AMBER or CHARMM force fields, where it reliably reproduces the electrostatic environment of kinase binding pockets.
Ensemble Strategy
To avoid the well-recognised risk of a single MD trajectory becoming trapped in a minor conformational sub-state, an ensemble approach was employed:
1. Three independent 10 ns replicate simulations, each initialised with a distinct set of randomised atomic velocities, were executed to sample divergent thermodynamic pathways and verify reproducibility of the binding pose.
2. One extended 100 ns production simulation was conducted to probe the long-term kinetic stability of the complex, including any slow-onset conformational transitions or delayed dissociation events that fall outside the window of shorter simulations.
All simulations were run following standard preparation: the protein–ligand complex was placed in a periodic boundary box, solvated with TIP3P water molecules, charge-neutralised with counter-ions, energy-minimised to remove steric clashes, and thermodynamically equilibrated to physiological conditions (300 K, 1 atm).
RMSD Analysis and Thermodynamic Stability
Root Mean Square Deviation (RMSD) the average atomic displacement relative to the initial reference structure was used to quantify both global protein stability and local ligand mobility throughout the 100 ns trajectory.
The protein backbone RMSD stabilised and plateaued at approximately 3.0–4.0 Å (0.3–0.4 nm) within the first tens of nanoseconds. This magnitude of fluctuation is expected and physically meaningful for a multidomain kinase: proteins are not rigid crystals, and the conformational breathing required for function is captured here as it would be in a real biological setting. The plateau rather than continued drift confirms that the simulated EGFR T790M structure maintained overall structural integrity throughout the run.
More critically, the ligand heavy-atom RMSD remained tightly constrained throughout the simulation. A sustained ligand RMSD below 2.0–3.0 Å is the widely accepted criterion for a stable, pharmacologically viable binding pose in MD-based drug design. A molecule with poor complementarity to its binding site will rapidly destabilise, typically producing RMSD excursions exceeding 5.0 Å as it tumbles into the bulk solvent. No such behaviour was observed for the OV4 lead compound.
Global thermodynamic indicators were equally reassuring: potential energy stabilised around −936,000 kJ/mol and kinetic energy held steady at approximately 204,800 kJ/mol across the entire trajectory, with no evidence of sudden steric clashes, energetic discontinuities, or partial unfolding events.
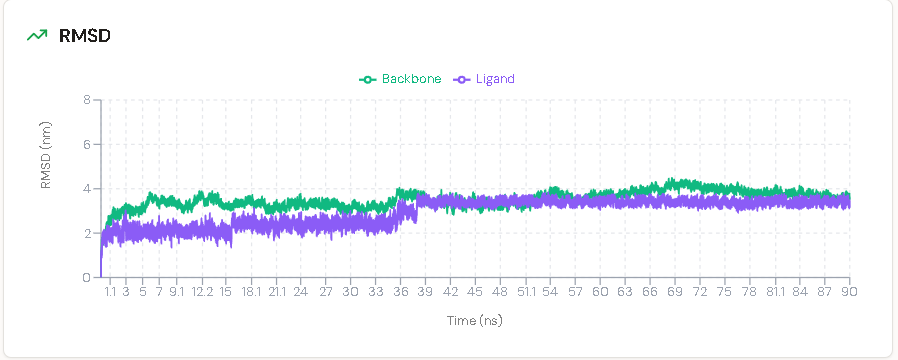
Interpreting the 35 ns Structural Transition
The most informative event in the extended trajectory was a discrete upward shift in ligand RMSD occurring at approximately 35 ns, at which point the ligand deviation rose to parallel the protein backbone at approximately 3.5 Å.
A superficial reading of this event might suggest ligand instability. The extended 100 ns window provided the context necessary to interpret it correctly. EGFR and other kinases are intrinsically plastic macromolecules: over extended nanosecond timescales it is routine for local side-chain rotamer flips, activation loop breathing motions, or solvent network reorganisations to occur in response to a bound ligand a phenomenon known as induced fit. Critically, following the 35 ns positional translation, the ligand RMSD did not continue to rise. Instead, it immediately established a new horizontal plateau at approximately 3.5 Å that persisted for the remaining 65 ns of the trajectory.
This behaviour is diagnostic of induced-fit accommodation rather than incipient dissociation. Had the shift reflected unfavourable binding energetics, the RMSD would have continued an unabated upward trajectory towards solvent escape. Instead, the system transitioned into a slightly reorganised thermodynamic equilibrium, confirming that the pre-covalent complex is robust against the receptor's natural conformational dynamics.
Interaction Fingerprints: Confirming Pre-Covalent Geometry
Two additional metrics from the 100 ns trajectory directly support the molecule's readiness for covalent chemistry:
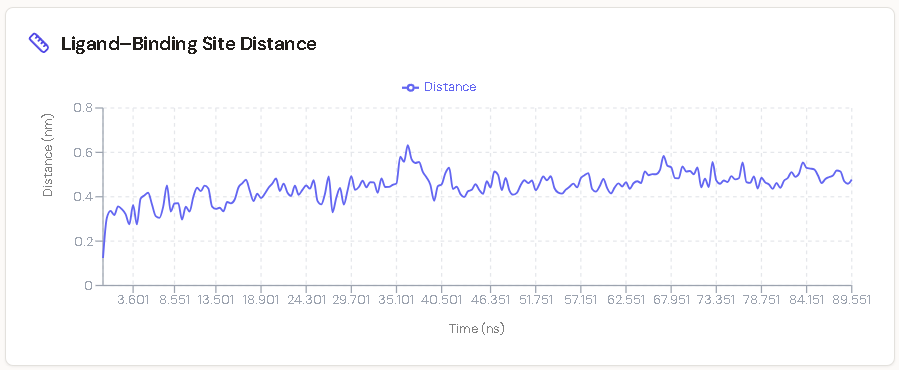
1. Ligand–Binding Site Distance
The distance between the molecule and the core binding site fluctuated within a constrained range of 0.4–0.5 nm (4–5 Å) throughout the full trajectory. Successful Michael addition requires the acrylamide β-carbon to approach within approximately 3.6–4.0 Å of the Cys797 sulfur. The MD data confirm that the ligand does not drift away from the reactive zone at any point during the simulation.
2. Sustained Hydrogen Bond Network
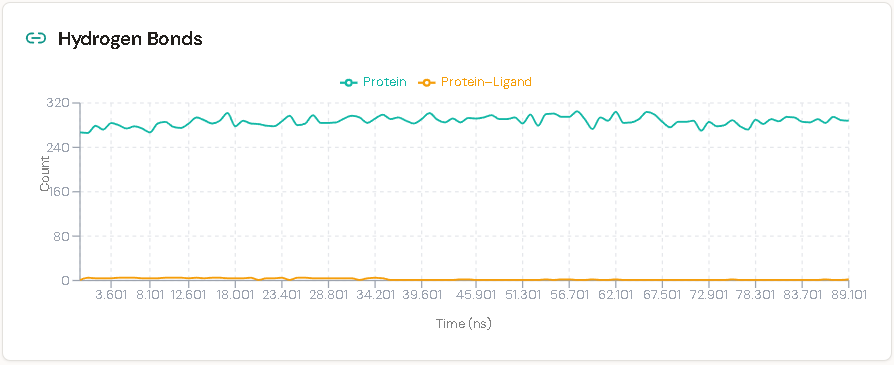
The ligand maintained an uninterrupted network of three hydrogen bonds with the surrounding pocket residues across the entire 100 ns run. While the broader protein structure naturally flexed sustaining approximately 280–300 internal hydrogen bonds with normal fluctuation the three ligand-specific anchor contacts never broke. This persistent electrostatic tethering provides the geometric constraint necessary for the acrylamide warhead to remain properly oriented relative to Cys797.
Summary and Comparative Context
The table below places the designed OV4 lead compound in context relative to established inhibitors and the initial de novo scaffold from which it was derived
| Molecule | Binding | Score | Strength | Limitation |
|---|---|---|---|---|
| Gefitinib | Reversible | ~−6 to −7 | Hinge binding (Met793) | Clash with Met790 |
| Osimertinib | Covalent (Cys797) | ~−8 to −9 | Mutant selective | C797S resistance |
| mol_99 | Non-covalent | −8.18 | Stable pocket fit | No covalent action |
| OV4 (lead) | Covalent-ready | −9+ | Fits Met790 + aligns Cys797 | Needs validation |
Conclusions
The results of this computational study support three main conclusions:
The LiteFold de novo design module is capable of generating acrylamide-warhead inhibitor candidates with docking profiles comparable to established third-generation EGFR inhibitors, without starting from a known scaffold.
The mol_99/OV4 lead compound maintains a stable pre-covalent binding state under dynamic conditions across a 100 ns MD trajectory, as evidenced by constrained ligand RMSD, persistent hydrogen bonding, and sustained proximity to Cys797. The 35 ns structural transition is consistent with induced-fit pocket accommodation rather than dissociation.
The integration of AI-guided de novo generation with MD-based dynamic validation represents a meaningful advance over static docking workflows for covalent inhibitor design, where pre-covalent geometric fidelity is a mandatory prerequisite for effective warhead chemistry.
These computational findings establish a strong rationale for progression to experimental validation including synthesis of the OV4 candidate, biochemical Ki and kinact/Ki determination, and cellular potency profiling in T790M-positive NSCLC models. The extent to which computationally predicted binding geometries translate to measured inhibitory activity will be the definitive test of this workflow.