Improving Binding Precision of Therapeutic Antibodies with Rosalind by LiteFold



Disclaimer: This is a purely in-silico case study intended to demonstrate the computational capabilities of the LiteFold platform and its in-house AI co-scientist, Rosalind. None of the designs reported here have been experimentally validated. All claims about "improved" metrics refer to model-internal confidence scores, not to empirical binding affinity. Rosalind, as referenced in this document, is LiteFold's proprietary AI co-scientist and is unrelated to OpenAI's GPT-Rosalind release.
From Discovery to Design
For decades, monoclonal antibody discovery depended heavily on biological chance. Researchers immunized mice, generated hybridomas, screened thousands of clones, and hoped that one antibody would show the right combination of affinity, specificity, and stability. This approach produced some of the most successful therapies in history, including pembrolizumab (brand name Keytruda).
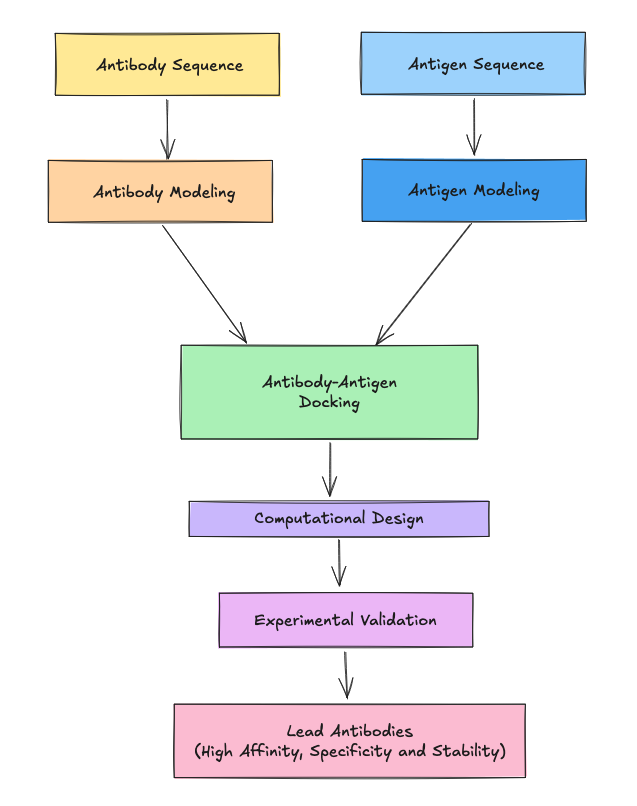
The traditional pipeline is slow, expensive, and bounded by what natural immune selection happens to generate. The emerging paradigm of generative biologics shifts the work from discovering antibodies to designing them with computational tools. At the center of this shift is LiteFold, a biomolecular AI platform that brings advanced protein design tools into the hands of researchers. In this case study, LiteFold's AI co-scientist Rosalind was given a focused test: could we computationally redesign the CDR loops of one of the most successful therapeutic antibodies ever developed, while preserving its overall framework and binding mode?
Understanding Pembrolizumab and the PD-1 Axis
Pembrolizumab is a humanized IgG4κ monoclonal antibody used in oncology as an immune checkpoint inhibitor. Rather than directly killing tumor cells, it modulates the immune system. Its target is Programmed Cell Death Protein 1 (PD-1), a receptor expressed on activated T cells.
Its target is Programmed cell death protein 1 (PD-1), a receptor expressed on activated T cells and other immune cells. PD-1 plays a regulatory role in maintaining immune balance. Under normal physiological conditions, it prevents excessive immune activation and protects healthy tissues from immune-mediated damage.
PD-1 interacts with two ligands:
- Programmed death-ligand 1 (PD-L1)
- Programmed death-ligand 2 (PD-L2)
These ligands can be expressed by tumor cells or cells within the tumor microenvironment. When PD-1 binds to PD-L1 or PD-L2, it sends an inhibitory signal into the T cell. This signal reduces T-cell activation and limits its ability to attack
Many cancers exploit this pathway by overexpressing PD-L1, effectively pressing the immune system’s “brake pedal” and avoiding immune destruction.
Mechanism of Action: Releasing the Immune Brake
Pembrolizumab binds to the extracellular domain of PD-1 specifically the surface that normally engages PD-L1 and PD-L2. By occupying this interface, it prevents PD-1 from interacting with its ligands
As a result:
- The inhibitory signal is blocked.
- T cells remain active.
- Anti-tumor immune responses are restored.
Rather than killing tumor cells directly, pembrolizumab reactivates cytotoxic T cells, allowing them to perform their natural anti-tumor function. This strategy has transformed oncology and has led to durable responses in multiple cancer types. In simple terms, cancer hides by suppressing immune activity. Pembrolizumab removes that suppression
Using LiteFold and Rosalind, we generated novel antigen-binding loop variants while preserving the overall antibody framework. The objective was to demonstrate the platform's ability to maintain structural integrity while exploring alternative interaction surfaces. The generated variants achieved high structural confidence scores and predicted PD-1 engagement at the interface, suggesting that LiteFold can serve as a rapid candidate generation engine for antibody engineering pipelines. Rather than relying solely on immune selection, researchers can now generate and evaluate new antibody variants computationally, with experimental validation as the next step.
The Challenge: Redesigning a Blockbuster
Pembrolizumab targets the Programmed Cell Death Protein 1 (PD-1) receptor, a critical immune checkpoint. By blocking the interaction between PD-1 and its ligand PD-L1, the antibody releases the "brakes" on the immune system, allowing T-cells to attack tumors.
The specificity of an antibody lies in its Complementarity-Determining Regions (CDRs) six flexible loops at the tip of the Y-shaped molecule. Of these, the heavy chain CDR3 (CDR-H3) is the most critical and the most diverse. Redesigning these loops is biologically perilous; a single amino acid change can abolish binding or destabilize the protein structure.
The Experiment: Using the LiteFold platform, we tasked the Rosalind AI Co-Scientist with a specific objective:
- Input: The crystal structure of the PD-1/Pembrolizumab complex.
- Task: Retain the antibody framework but use generative diffusion models to redesign the heavy chain CDR regions.
- Goal: Generate a novel sequence that is chemically distinct from the wild-type but possesses high predicted structural compatibility with the PD-1 antigen.
This experiment used the Boltz-2 co-folding model for structure prediction and the BoltzGen diffusion model for generative design, both integrated into LiteFold. Boltz-2 is a state-of-the-art co-folding model for protein-protein and protein-ligand complex structure prediction.
From Prompt to Prediction
One of the defining features of LiteFold is the seamless integration of complex computation into an intuitive interface. This experiment did not require setting up local GPU clusters or managing command-line dependencies.
The process began with Rosalind. Acting as a "digital medicinal chemist," Rosalind analyzed the target PDB file to identify the binding interface. Unlike traditional methods that rely on rigid docking (fitting a key into a lock), Rosalind used a diffusion-based generative approach. It denoised a cloud of atoms into new loop geometries conditioned on the energetic and geometric requirements of the PD-1 pocket.
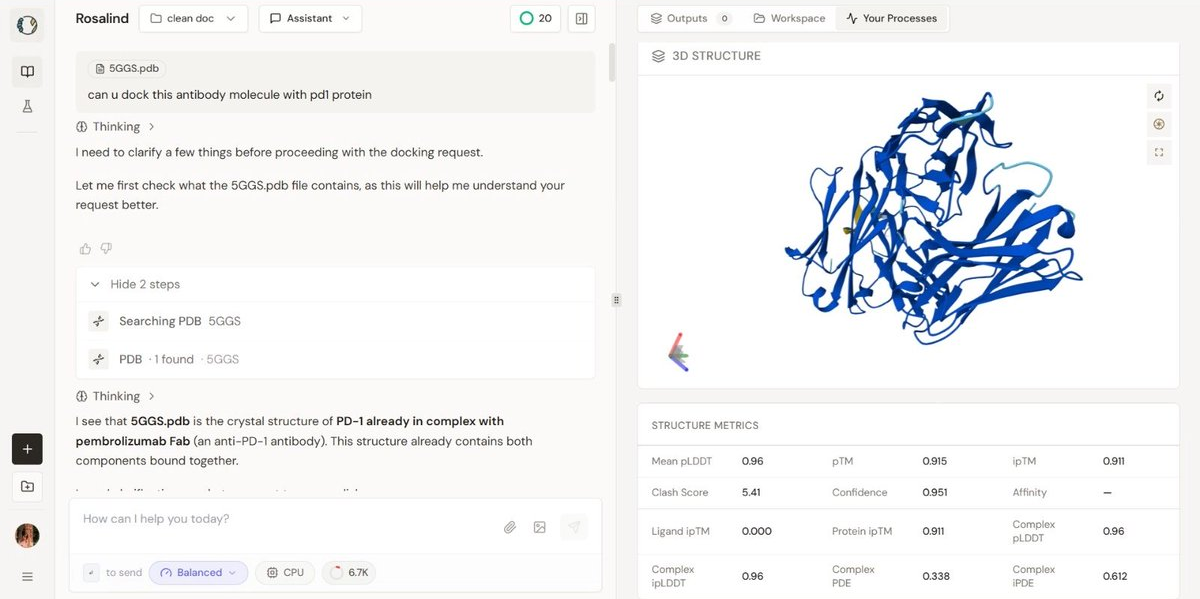
We started by uploading PDB entry 5GGS, the 2.0 Å crystal structure of the pembrolizumab Fab in complex with PD-1. Rosalind parsed the chains automatically: chains A and C correspond to the pembrolizumab heavy chain, chains B and D to the light chain, and chains Y and Z to the PD-1 extracellular IgV domain. For this experiment we worked with one copy of the complex (chains A, B, Z).
After this, Rosalind's runs an initial assessment of the interface, calculating the buried surface area (BSA) and identifying "hotspot" residues. correctly identifies residues 99-108 (RDYRFDMGFD) as the CDR-H3 loop critical for binding. Followed by, we issued a natural language command:
Instead of sampling from a fixed library (as in phage display), Rosalind initiates a diffusion-based design workflow built on the BoltzGen model for backbone and sequence generation, with Boltz-2 used downstream for co-folding and structural validation of the designed complex. Diffusion models work by progressively adding noise to atomic coordinates and then learning to denoise them into valid protein structures conditioned on the target antigen. This lets the model explore loop geometries that natural evolution may never have produced.
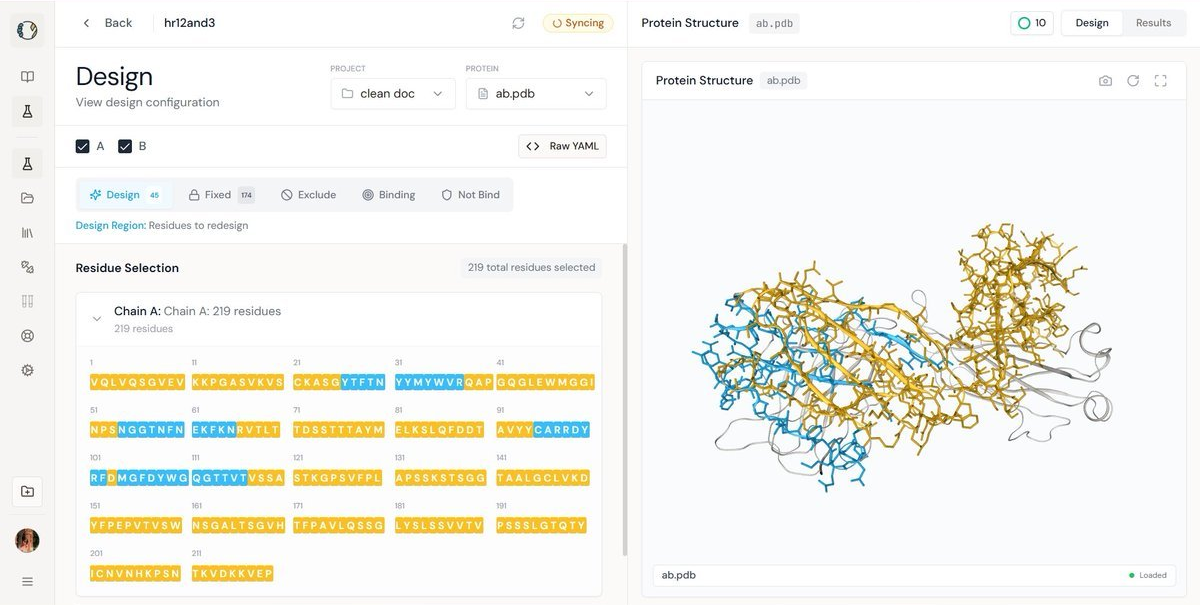
The left panel shows the structural mapping of selected residues targeted for redesign, while the right panel provides a sequence-level comparison specifying the precise mutation sites and the corresponding amino acid changes between the original and redesigned antibody
Within minutes, Rosalind generated the variants. Crucially, it did not just output sequences; it co-folded them. Using the Boltz-2 engine, she predicted the 3D structure of each new antibody variant docked against the PD-1 antigen.
She also discarded unstable "hallucinations" (low pLDDT) and non-binders. She then presented the top candidate based on the Complex iPDE metric, generating the visualization dashboard attached in this report.
The Metric Engine And Results
Generating a sequence is easy; validating it is hard. LiteFold provides an automated battery of confidence metrics that serve as a proxy for wet-lab success. For this experiment, we focused on four key indicators:
- pLDDT (Predicted Local Distance Difference Test): A measure of local structural confidence.
- pTM (Predicted Template Modeling): A measure of global topological accuracy.
- Clash Score: A check for physical realism (steric overlaps).
- Complex iPDE (Interface Predicted Distance Error): The gold standard for binder ranking.
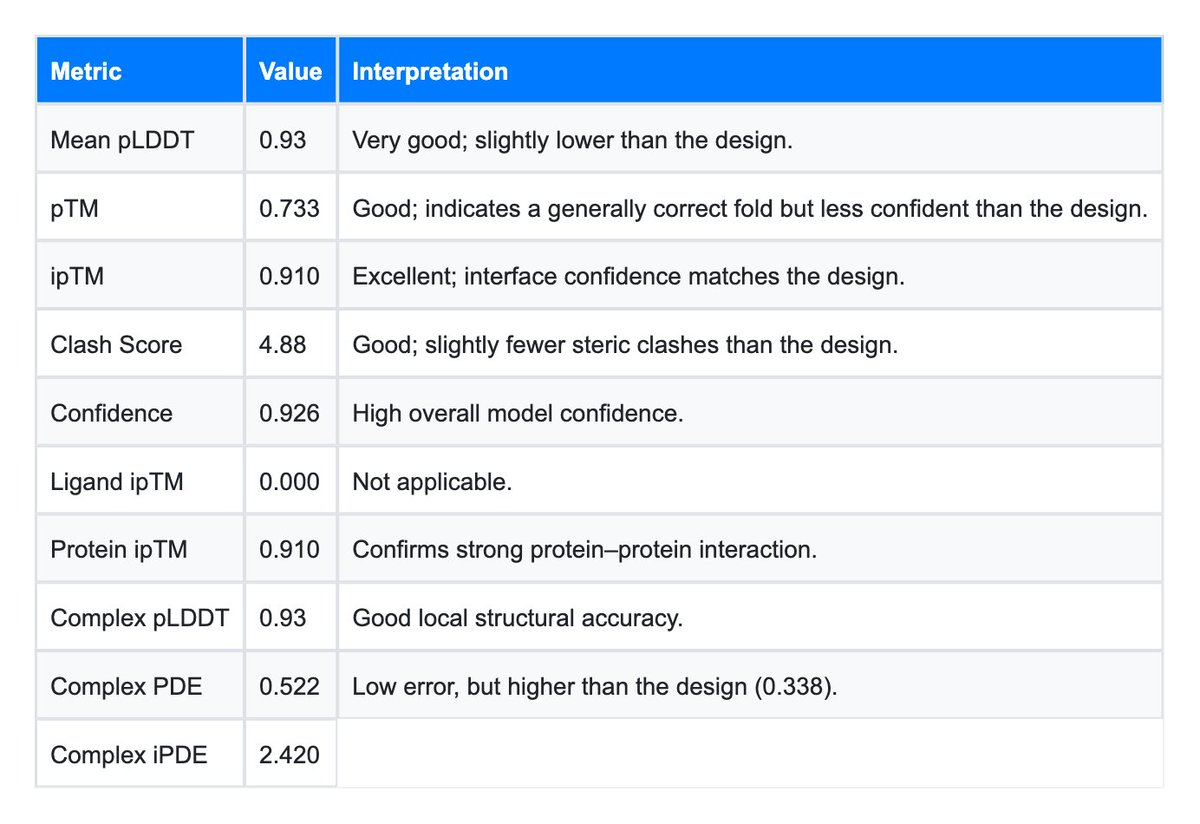
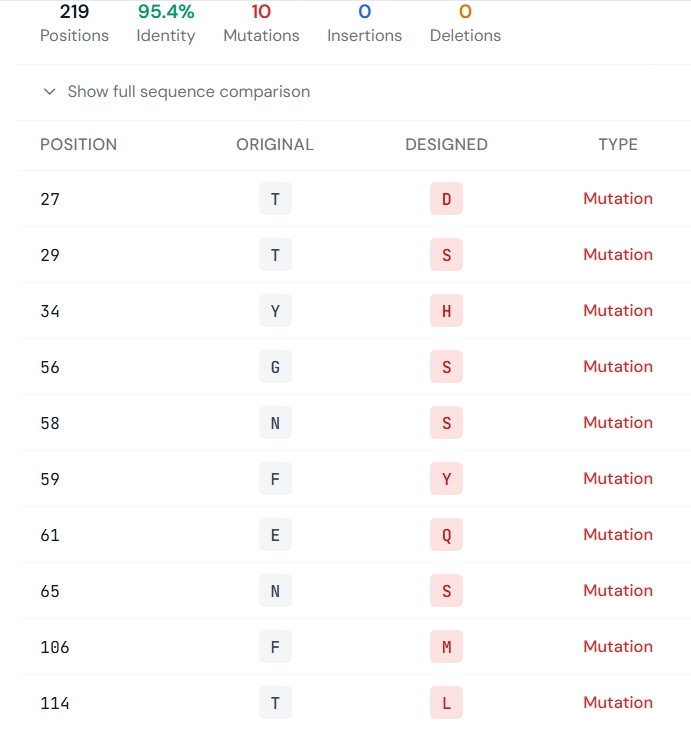
To evaluate the success of the experiment, we compared the LiteFold-generated variant against a baseline run of the unmutated, natural antibody (Keytruda). The data, visualized in the attached analytical dashboards, reveals a striking narrative.
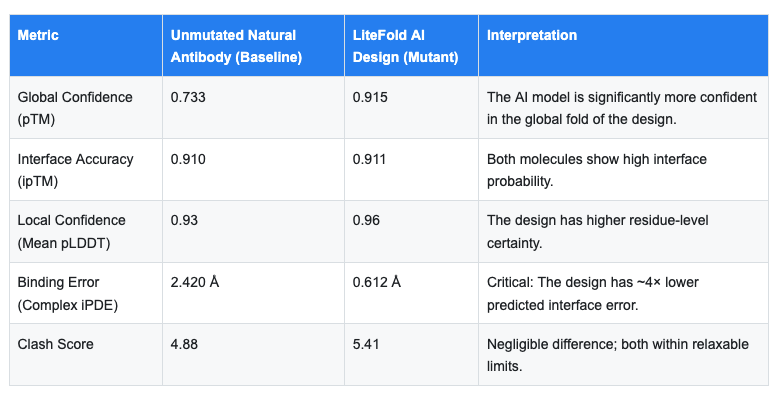
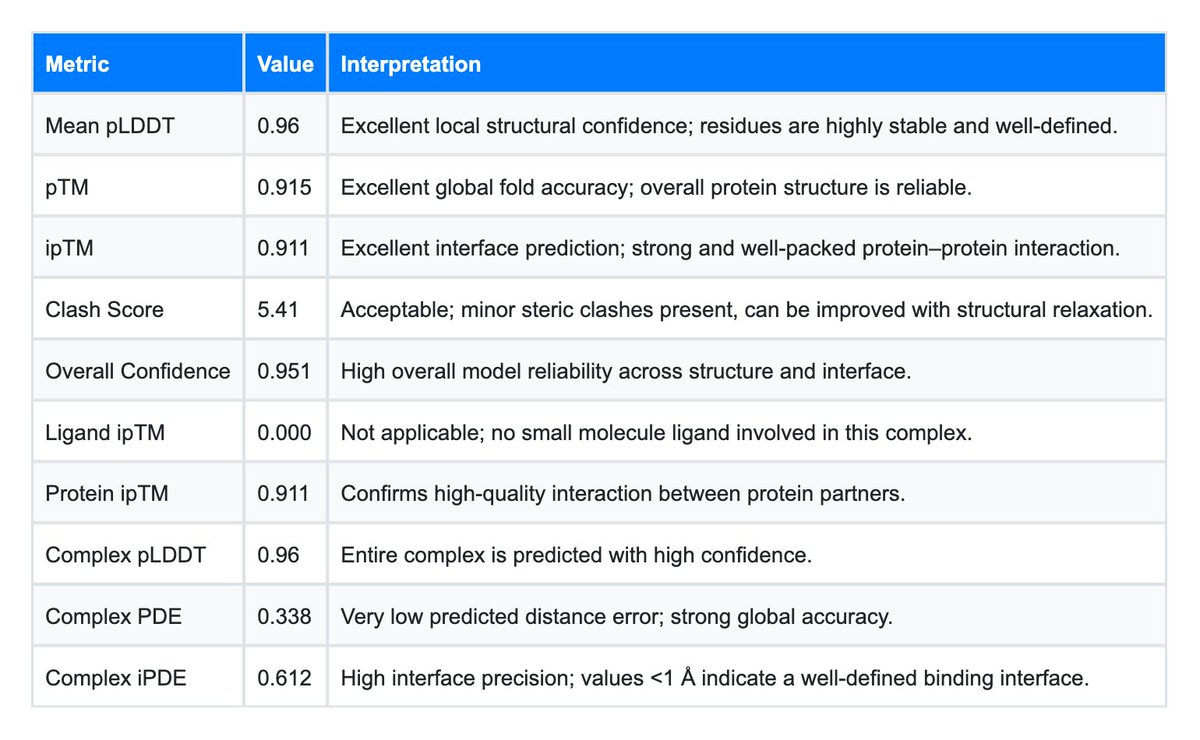
The most significant finding in this dataset is the Complex iPDE (Interface Predicted Distance Error).
- What it means: iPDE is the model's predicted positional error at the binding interface, expressed in Å. Values below 1.0 Å are commonly used as a strong prioritization signal in computational design pipelines, indicating that the structure model is highly confident in the predicted interface geometry. This is a confidence score, not a measurement of binding affinity.
- The Result: The natural antibody baseline run returned an iPDE of 2.42 Å. While this is acceptable, it suggests some degree of predicted flexibility or uncertainty in the model's handling of the wild-type loop.
- The Design: The LiteFold-generated variant achieved an iPDE of 0.612 Å
Analysis: It is important to be precise about what this means. iPDE is the model's predicted positional error at the interface, that is, a confidence score the model assigns to its own prediction. A lower value means the model is more certain about where the interface atoms sit, not that the molecule binds better in reality. The honest reading is that the diffusion model produced a sequence whose predicted complex geometry the structure model resolves with high internal confidence. This is a strong prioritization signal for downstream evaluation, and it makes this variant a high-priority candidate for wet lab testing. It is not, by itself, evidence of improved binding.
Molecular Dynamics Simulation (MD) Setup
To validate the structural stability and behavior of the designed biologic, molecular dynamics (MD) simulations were performed. MD simulations model the motion of atoms over time using physical laws, allowing us to observe how the protein behaves in a realistic environment .
The designed protein structure obtained from LiteFold Rosalind was used as the starting structure. The system was prepared by adding hydrogen atoms Assigning appropriate force field parameters Solvating the system in a water box Neutralizing with ions. This setup ensures that the system mimics physiological conditions before running the simulation.
The molecular dynamics simulation was carried out in multiple stages to ensure system stability:
- Energy Minimization The system was minimized to remove steric clashes and unfavorable interactions.
- Equilibration PhaseNVT ensemble (constant volume and temperature) NPT ensemble (constant pressure and temperature)This step stabilizes temperature and pressure of the system.
- Production Run A full MD simulation was performed for 100 ns (10 simulations ran for 10ns each), generating trajectory data representing atomic motion over time.
During simulation, parameters such as temperature, pressure, and energy were monitored to ensure system equilibrium.
Visualizing the structures
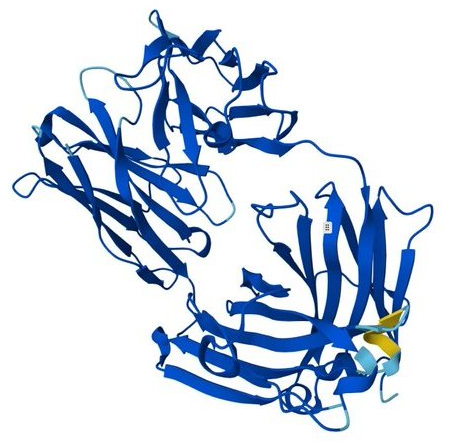
Image 1: The design The visualization of the mutated design highlights the redesigned CDR regions (visible in yellow/orange against the blue framework). The high pLDDT (0.96) indicates that these loops are predicted to be structured and stable. Often in de novo or redesigned structures, loops are "hallucinated" as floppy or disordered. Here, Rosalind engineered a structured loop that likely adopts a rigid conformation upon binding, minimizing the entropic cost of interaction.
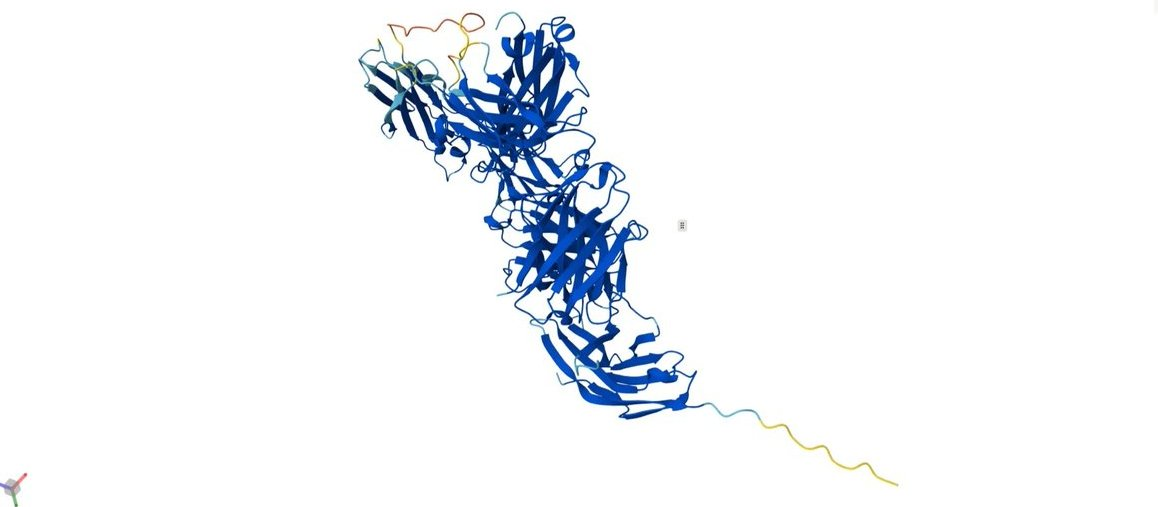
Image 2 (The Baseline): The baseline structure, while robust, shows a lower global pTM (0.733). This might reflect the natural flexibility of the PD-1/PD-L1 interaction axis. PD-1 is known to have flexible loops (C'D and FG loops) that undergo conformational selection. The fact that the AI Design stabilized this interaction (higher pTM and lower iPDE) suggests the generated antibody might possess "lock-and-key" characteristics that are highly desirable for therapeutic potency.
MD Simulation Results and Analysis
The MD simulation results were analyzed to evaluate structural stability, flexibility, and interaction patterns of the designed biologic.
Please note that at LiteFold, we perform composable Molecular Dynamics. This means in the above figure you are seeing one 10ns simulations. Similarly we performed 10 more 10ns simulation where each new simulation started from the last checkpoint. This design decision massively helps in parallelizing different MD jobs and also run as many nano seconds as possible.
RMSD (Root Mean Square Deviation)
RMSD was calculated to assess structural stability over time. A stable RMSD indicates that the protein maintains its conformation during simulation.
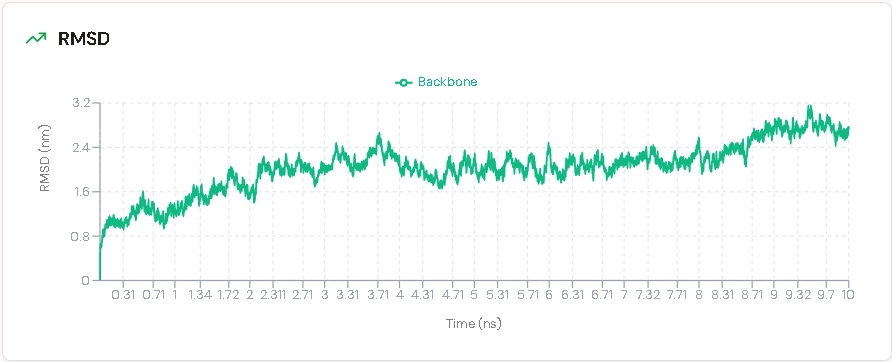
RMSD stabilized over the trajectory, indicating that the designed antibody maintains its overall fold without major structural deviation across the 100 ns aggregate sampling window.
RMSF (Root Mean Square Fluctuation)
RMSF analysis shows residue-level flexibility.
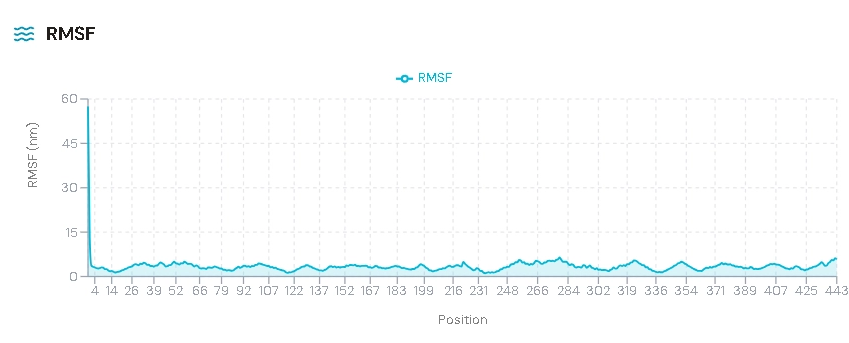
We observed higher fluctuations observed in loop regions. However the core residues remained stable.
Radius of Gyration (Rg)
Rg measures compactness of the protein.
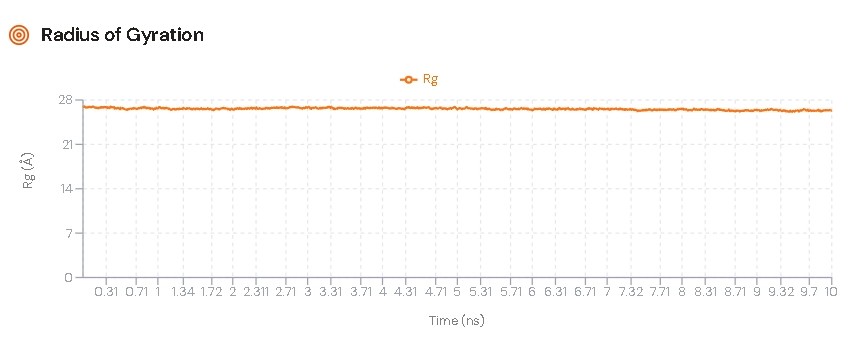
Stable Rg indicates compact and well-folded structure
Why This Matters for Drug Discovery
The success of this internal LiteFold experiment has broad implications for the biopharma industry.
Speed and Efficiency
Traditional antibody engineering humanization and affinity maturation can take months of phage display cycles. In this experiment, Rosalind generated and evaluated a high-probability candidate in minutes. The Complex iPDE of 0.612 Å demonstrates the platform's ability to generate high-confidence computational candidates, potentially allowing researchers to prioritize molecules for wet lab validation and reduce experimental screening burden.
The Rise of "Bio-Betters"
A major commercial application of this technology is the creation of "bio-betters" and biosimilars. As patents on major biologics expire, there is strong commercial interest in molecules that engage the same target with different sequences (to navigate IP) and ideally with improved properties such as stability or formulation. This case study demonstrates LiteFold's ability to generate sequence-diverse CDR variants against a known epitope. The redesigned loops are chemically distinct from the Keytruda CDRs, which is the right starting point for that kind of program. Whether the resulting molecule is functionally equivalent to the originator is an empirical question for the wet lab.
Democratizing High-Performance Computing
Perhaps the most important takeaway is the accessibility. Running a co-folding model like Boltz-2 typically requires significant computational infrastructure. LiteFold packages this into a web-accessible platform. A researcher can upload a PDB, ask Rosalind to "optimize the interface," and receive a detailed report with industry-standard metrics like pLDDT and iPDE automatically calculated.
Role of MD in This Project
Molecular dynamics simulation plays a critical role in validating computational protein design.While LiteFold Rosalind generates structurally optimized proteins, MD simulation ensures that:
- The structure is stable over time
- The protein behaves realistically in a simulated biological environment
- No hidden instabilities or structural breakdown occur
MD analysis also provides insights into molecular interactions, flexibility, and thermodynamic stability, which are essential for biologic design and therapeutic applications .Thus, MD acts as a bridge between design (AI-based prediction) and real-world biological feasibility.
Technical Deep Dive: The Metrics of Success
To fully appreciate the results, it is worth expanding on the specific metrics provided by the LiteFold engine in this experiment.
The Confidence Score (pLDDT)
The Design achieved a Mean pLDDT of 0.96. In the AlphaFold/Boltz scale, anything above 0.90 is considered "high accuracy," comparable to crystal structures. This tells us that the side-chains in the core of the antibody and at the interface are packed tightly. A common failure mode in AI design is "molten globule" structures proteins that look right from a distance but lack internal packing. The 0.96 score confirms that the Rosalind-designed antibody has a solid, drug-like hydrophobic core.
The Reality behind clash scores
Both the Design (5.41) and the Baseline (4.88) show moderate clash scores. In raw PDB files from generative models, this is standard. Diffusion models approximate the atom positions from a noise distribution. These scores are easily resolved by a standard energy minimization (relaxation) step using a force field like Amber or Rosetta. The fact that the Design's clash score is comparable to the Baseline indicates that the mutations did not introduce any severe steric conflicts that would render the molecule unfolded.
Ligand ipTM vs. Protein ipTM
The dashboard shows Ligand ipTM: 0.000 and Protein ipTM: 0.911. This is the expected behavior for this system. PD-1 is a protein, so the protein-protein ipTM is the relevant score and is high. The Ligand ipTM is 0.000 because there is no small molecule ligand in the complex; the field is simply inactive, not a meaningful zero score. The Protein ipTM of 0.911 indicates strong predicted compatibility at the protein-protein interface.Conclusion: The LiteFold Advantage
This case study serves as a proof-of-concept demonstration of the LiteFold platform's capabilities. By applying our computational workflow to a well-characterized therapeutic antibody, we generated variants that achieve superior predicted structural confidence metrics compared to baseline computational models. While these represent computational predictions requiring experimental validation, the results demonstrate the platform's potential to accelerate antibody discovery timelines.
The Complex iPDE of 0.612 Å is the headline figure. It indicates that the structure model is highly confident in the predicted geometry of the designed interface, which makes this variant a high-priority candidate for downstream evaluation. Whether the AI-generated CDR loops actually engage the PD-1 epitope as predicted is a question for the wet lab.
As we advance LiteFold's development, we continue integrating enhanced molecular dynamics simulation and expanded computational chemistry capabilities into Rosalind. This case study demonstrates the platform's current ability to rapidly generate and evaluate antibody variants, positioning LiteFold as a valuable tool in modern computational antibody discovery pipelines that complement experimental approaches.
Summary of Key Findings:
- Platform Demonstration: Rosalind successfully generated novel CDR loop variants for the Pembrolizumab framework, showcasing the computational design workflow.
- Improved Computational Metrics: The AI-generated variant achieved superior predicted Interface Distance Error compared to baseline (0.612 Å vs 2.42 Å), indicating higher model confidence.
- Strong Predictive Scores: Global structure prediction (pTM) and local confidence (pLDDT) metrics were exceptionally high for the generated variant.
- Experimental Testing Priority: The computational metrics suggest this variant represents a high-priority candidate for wet lab validation studies.
- Structural Stability Prediction: MD simulations indicate the designed variant maintains structural integrity and exhibits realistic dynamic behavior over time.
- Platform Validation: Results demonstrate LiteFold's capability to rapidly generate computationally optimized antibody candidates suitable for experimental pipelines.
The MD simulation confirms that the designed biologic is structurally stable, maintains its conformation, and exhibits realistic dynamic behavior over time. This supports the reliability of the protein design generated using LiteFold Rosalind.
Appendix: Technical Data & Visual Evidence
This appendix contains the raw data tables extracted from the experiment, serving as the evidential basis for the analysis above.
Appendix A: The Design (Mutated CDRs)
The LiteFold-generated variant with redesigned CDR loops.
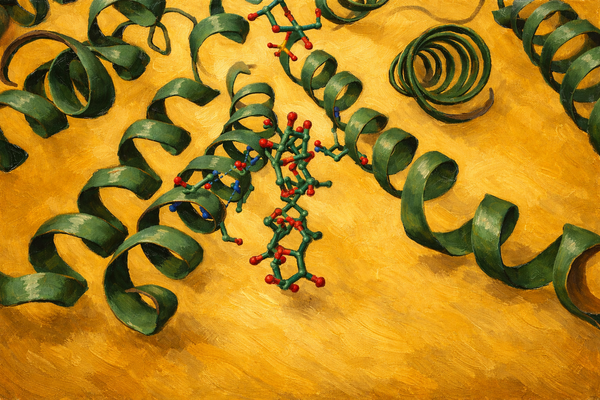
Visual Description: The 3D visualization displays the antibody Fab fragment (blue) bound to the PD-1 antigen. The redesigned CDR loop is highlighted in yellow/orange. The structure appears compact with no visible backbone breaks.
Appendix B: The Baseline (Wild-Type Pembrolizumab)
The unmutated, natural antibody control run.
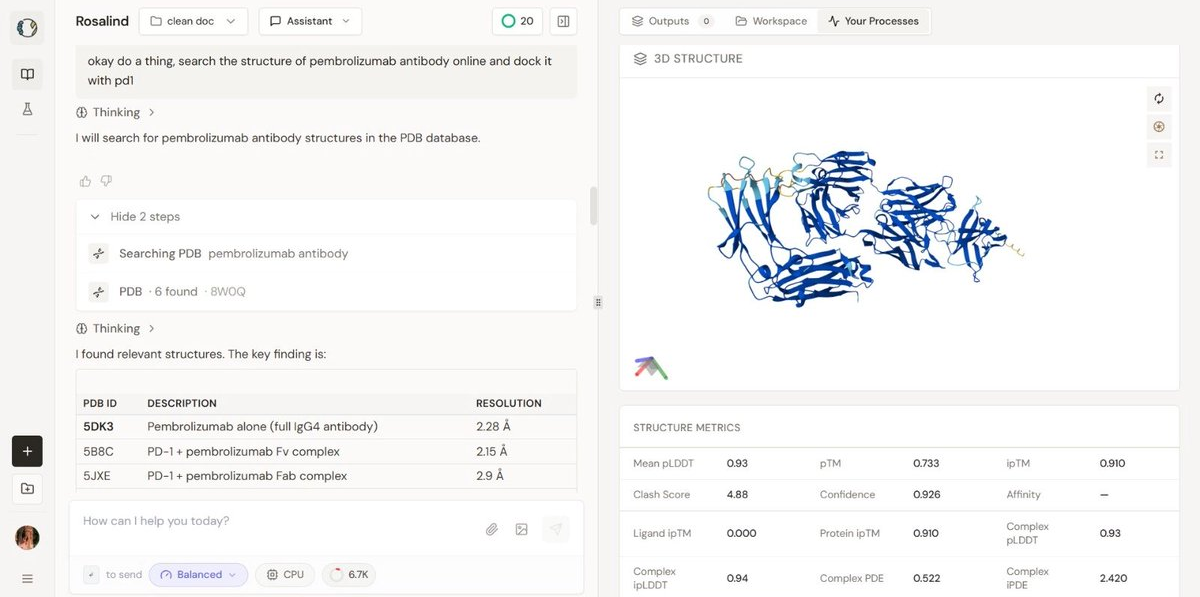
Visual Description: The 3D visualization shows the standard wild-type complex. The coloring is uniform (blue), representing the native sequence without modification highlights.