Fail Fast, Fail Cheap: In-Silico Toxicology Pipelines for Early Drug Candidate


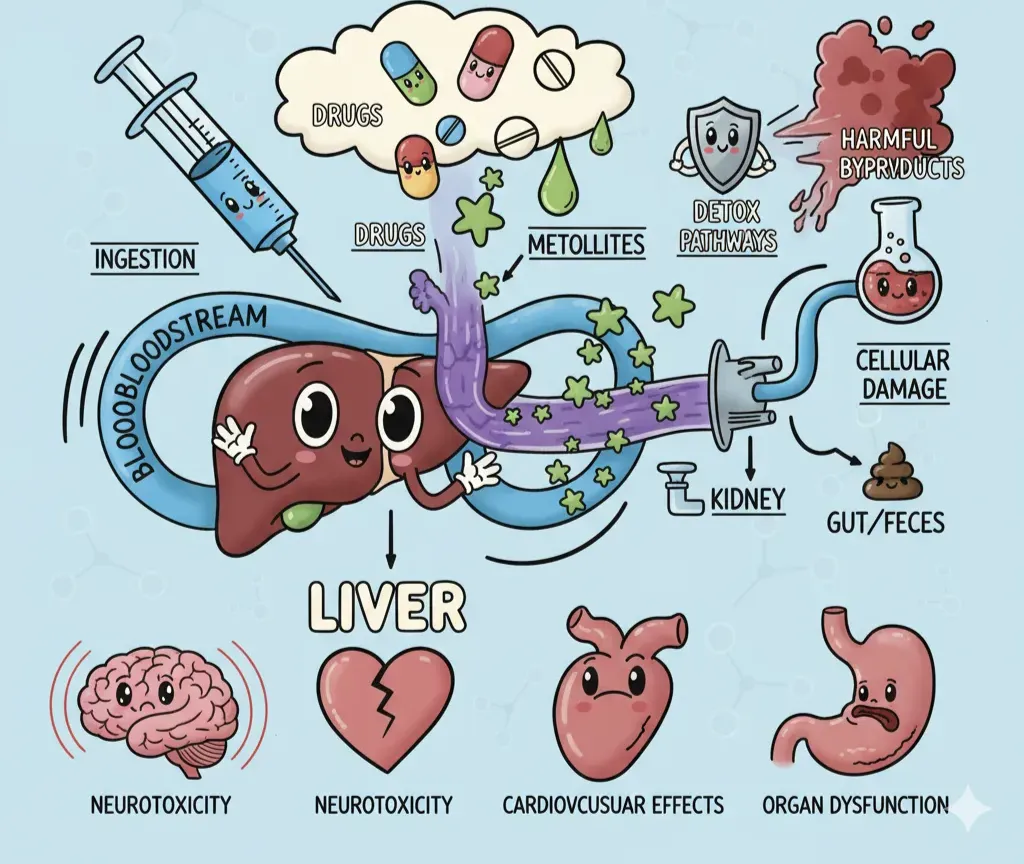
Drug discovery is basically a casino where the house almost always wins. Around more than 90% of drug projects never make it to patients, and the price of failure climbs steeply the further along you go. Flop early in discovery? That’s about a million dollars down the drain. Flop in late-stage clinical trials? That’s a jaw-dropping $2.6 billion burned.
One of the biggest culprits behind these late-stage wipe outs is toxicity, drugs that look promising but turn out to be harmful once tested in humans. Toxicity accounts for nearly a third of candidate terminations. For a well-funded pharma giant, that’s painful. For a startup? It’s lethal.
Why All Toxicities Aren’t the Same
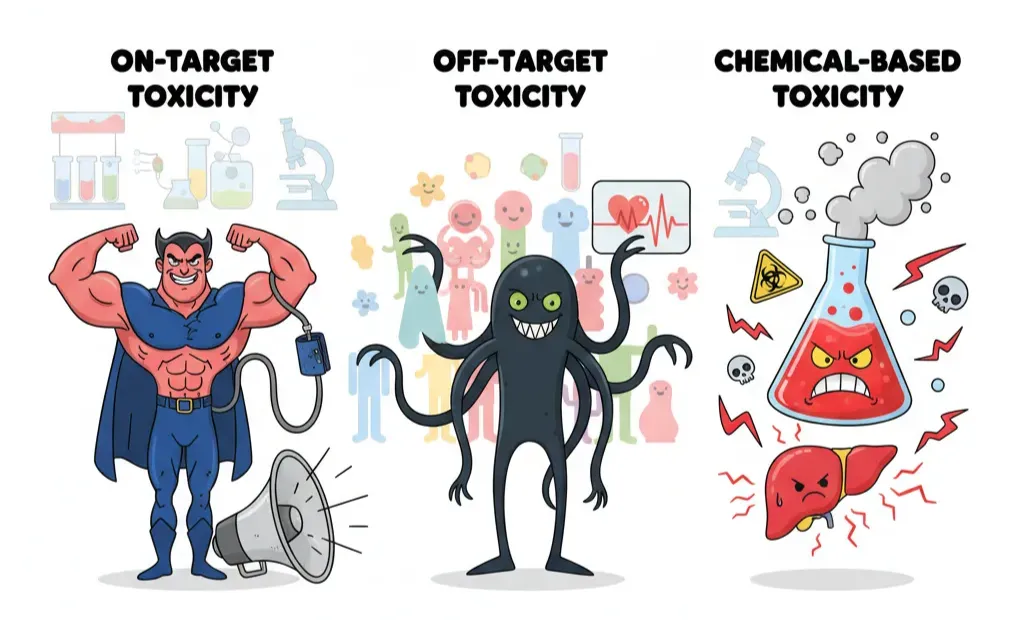
“Toxicity” sounds like a single villain, but in drug development it shows up wearing different masks. If you want to catch it early, you need to know which version you’re fighting. Broadly, toxic effects fall into three buckets:
- On-target toxicity – the drug is hitting exactly what it was designed to, but too hard. The biology is right, the dose is wrong. Blood pressure drugs that drop pressure so low you faint are a classic example. It’s the pharmacological equivalent of enjoying loud music until the speakers blow.
- Off-target toxicity – here the drug starts flirting with proteins it was never meant to bind. Kinase inhibitors are notorious: one was designed for a single kinase, but ended up interacting with dozens. Sometimes this broad activity helps (blocking cancer escape routes), other times it’s deadly (triggering arrhythmias through hERG channel binding).
- Chemical-based toxicity – some compounds don’t even need proteins to wreak havoc. They’re chemically unstable or reactive, forming toxic metabolites or generating free radicals. Acetaminophen overdose frying the liver is a textbook case.
Understanding which bucket a toxic effect falls into isn’t trivia, it shapes how you predict it, test it, and engineer around it.
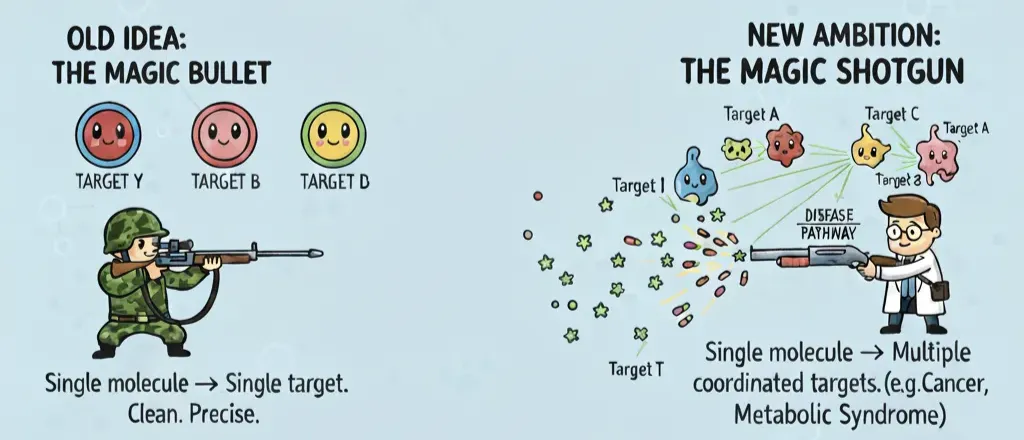
The Death of the Magic Bullet
For most of the last century, drug discovery worshipped the magic bullet idea: a single, exquisitely selective molecule that hits one target and leaves everything else alone. Clean, precise, safe.
Reality check: molecules are promiscuous. On average, a small-molecule drug binds between six and eleven different proteins. This “polypharmacology” used to be considered sloppy chemistry. Now it’s seen as the natural state of drug action—and sometimes, a feature.
Which is why the mindset has shifted from snipers to shotguns. The new ambition is a magic shotgun: a drug rationally designed to hit multiple targets in a coordinated way. In diseases like cancer, Alzheimer’s, or metabolic syndrome, nudging several nodes in a pathway often works better than hammering one in isolation. And from a practical angle, a single multi-target drug can also reduce pill burden, simplify pharmacokinetics, and cut down drug–drug interactions compared to combination therapy.
This is where computational toxicology earns its keep. Predictive models spit out results like “Molecule X binds Target Y”. On their own, those predictions are meaningless. The value comes from how you interpret them.
- If Target Y is the hERG potassium channel (the classic culprit behind drug-induced arrhythmias), then binding is a red flag: a signal your compound could be cardiotoxic.
- If Target Y is a receptor in an inflammatory pathway linked to another disease, the exact same prediction is a green light: an unexpected chance to repurpose the compound for a new indication.
That’s the trick: the computational output is neutral. It’s the biological context, what that protein does in health and disease, that flips it from liability to opportunity.
This is why drug repurposing has shifted from serendipity to strategy. Aspirin started as a painkiller and became a cornerstone of cardiovascular prevention. Today, instead of waiting for lucky accidents, we use predictive models to systematically chart a molecule’s binding “constellation,” then sort the stars into two camps: hazards that demand caution, and potential guides toward new therapeutic directions.
We are on a mission to make molecular and structural biology related experiments easier than ever. Whether you are doing research on protein design, drug design or want to run and organize your experiments, LiteFold helps to manage that with ease. Try out, it's free.
A tour of computational predictive methods
Broadly, there are two methods, both perspectives are essential, and the best pipelines use them together.
Ligand-Based Methods: look at the molecule itself (the “key”)
These methods work under a simple but powerful rule of thumb: similar molecules tend to act in similar ways. If you don’t have the 3D structure of the protein, you let the drug’s structure do the talking.
A: Quantitative Structure–Activity Relationship (QSAR)
The process involves translating the 2D or 3D chemical structure of a molecule into a set of numerical features. You take features of the molecule, like molecular weight, lipophilicity (logP), or how many hydrogen bonds it can form and convert them into numbers called descriptors.
A statistical model, such as a regression or classification algorithm, is then trained on a dataset of molecules with known activities (e.g., toxic or non-toxic) to find a mathematical equation that links the descriptors to the activity. The resulting model, in its simplest form, looks like this
Activity=f(descriptors)+errorwhere f can be any regression or classification function, such as a linear model, nonlinear regression, or machine learning model and
error represents the difference between the observed experimental biological activity and the activity predicted by the model
Feed in enough data about known molecules (toxic or safe), and the model learns patterns. Once trained, it can spit out predictions for new compounds. Like predicting house prices, the quality of the answer depends entirely on the quality of your dataset.
B: Pharmacophore Modeling
While QSAR considers the molecule as a whole, pharmacophore modeling focuses on identifying the essential 3D arrangement of features required for a drug to interact with a target or the “essential features” that matter for binding: hydrogen bond donors/acceptors, hydrophobic spots, aromatic rings, charges. It’s a kind of 3D blueprint of what’s needed for activity.
You can then use this blueprint as a search query, scanning vast compound libraries for molecules that fit the pattern, even if they look nothing like the original.
Structure-Based Methods: look at the protein it might fit into (the “lock”).
If you’ve got a high-resolution protein structure (from X-ray crystallography or cryo-EM), you can get a lot more specific.
A: Molecular Docking
As explained earlier in my blog on docking, the software tries to “fit” a drug molecule into the binding pocket of the protein. It tests millions of poses, scoring each one to estimate how well it sticks. The result is not just a binding score but a visual snapshot of how the drug might interact at the atomic level.
B: Reverse Docking ("Target Fishing")
Flip the game: instead of many drugs against one protein, test one drug against thousands of proteins. The goal is to spot all the possible targets,both intended and unintended. This is how you can flag potential off-target toxicities or repurpose existing drugs.
One striking example: Researchers used reverse docking to show that PFAS, environmental contaminants, bind strongly to the folic acid receptor, which is crucial for brain development. This binding disrupts folate uptake, explaining the link between PFAS exposure and neurodevelopmental problems.
Table 1: Summarizes computational predictive methods
| Method Category | Core Principle | What You Need (Data) | Example Techniques | Pros | Cons |
| Ligand-Based | Similar molecules have similar activities. | A dataset of molecules with known activity data. | QSAR, Pharmacophore Modeling, 2D/3D Similarity | Doesn't require a 3D target structure. Fast for large libraries. | Less mechanistic insight. Can struggle with novel chemical scaffolds. |
| Structure-Based | A drug's activity depends on its 3D fit to a target. | A 3D structure of the target protein(s). | Molecular Docking, Reverse Docking | Provides detailed mechanistic insight into binding. | Requires a known 3D target structure. Computationally intensive |
No single method rules them all. Instead, computational scientists build workflows that:
- Use ligand-based methods when speed is needed or when protein structures are missing.
- Use structure-based methods when detailed mechanistic insight is critical.
- Integrate both into layered pipelines, cross-validating predictions for reliability.
But here’s the non-negotiable truth: your model is only as good as your input data. QSAR collapses if the training dataset is biased or messy. Docking results are meaningless if the protein structure is poor quality. A huge part of the field isn’t the glamorous modeling, but the grunt work of collecting, cleaning, and curating solid data. Garbage in, garbage out, it’s the universal law of computational toxicology.
The AI Revolution in Toxicology
QSAR and docking have been around since your professors were grad students, but in the last decade, toxicology got rocket fuel: artificial intelligence (AI). What used to be linear regressions and rule-based “if this, then that” systems is now a playground of neural nets, graph models, and enough acronyms to make you more confuse. At its core, AI changed toxicology in two big ways:
Old School: Expert Systems
Before AI hype, software relied on wisdom from chemists into codified ruled based systems. These tools run on curated libraries of structural alerts (a.k.a. toxicophores): chemical substructures repeatedly associated with toxic outcomes.
Some examples includes:
- Derek Nexus (Lhasa Ltd.) – Probably the most widely used. It flags toxicity risks across endpoints like mutagenicity, carcinogenicity, skin sensitization, and more. It’s trusted in pharma pipelines and even considered by regulators.
- Toxtree (Open source, JRC) – A free alternative that encodes decision trees of toxicophores. Great for academic use or early-stage projects.
- HazardExpert / OncoLogic – Older U.S. EPA and commercial tools that applied similar alert logic, especially around carcinogenicity.
Now the question is, what counts as a “structural alert”?
- Nitroaromatic groups – associated with mutagenicity (due to metabolic activation into reactive intermediates).
- Epoxides – reactive three-membered rings that can covalently bind DNA or proteins, leading to genotoxicity.
- Anilines – linked to methemoglobinemia (blood toxicity).
- Michael acceptors (α,β-unsaturated carbonyls) – electrophilic hotspots prone to react with nucleophiles in proteins → toxicity.
- Aromatic amines – long-known red flags for carcinogenic risk.
These systems flag and provide reasoning: “Compound contains an aromatic amine substructure. Literature links similar compounds to hepatotoxicity due to reactive metabolite formation.” That transparency is their superpower. Table 2: Strengths vs Weaknesses of ruled-based systems
| Aspect | Strengths | Weaknesses |
| Interpretability | Every alert has a documented rationale with traceable evidence | Static knowledge base limited to known mechanisms |
| Coverage | Spans multiple toxicity endpoints with minimal input requirements | Over-pessimistic predictions leading to false positives |
| Speed | Rapid analysis using only SMILES strings | Endpoint bias (missing complex toxicities like hERG cardiotoxicity) |
| Validation | Well-established usage in regulatory settings | Difficulty handling novel chemical structures |
| Integration | Easy to incorporate into existing screening workflows | Limited ability to consider complex biological contexts |
New School: Machine Learning Systems
If expert systems are rulebooks written by chemists, machine learning (ML) systems are data-hungry interns that figure out the rules on their own.
They rely on models that chew through huge datasets of molecules with measured properties, toxicity endpoints, ADME (Absorption, Distribution, Metabolism, Excretion) profiles, etc. and learn the patterns automatically. Popular Platforms and Tools:
- ADMET Predictor (Simulations Plus): One of the longest-standing commercial platforms. It bundles models for solubility, metabolism, toxicity, and pharmacokinetics. Widely used in pharma pipelines.
- ADMET-AI (recent, open-access): Web platform that runs ML/DL models for a wide range of ADMET endpoints. Think of it as a “plug-and-play” toxicology predictor.
- DeepTox (NIH Tox21 Challenge winner, 2014): A pioneering deep learning framework trained on >10k compounds across 12 toxicity endpoints. It showed that neural networks could outperform traditional QSAR methods.
- ProTox-III: A webserver trained on massive datasets, predicts acute toxicity (LD50), hepatotoxicity, carcinogenicity, and more, with probability scores.
- ChemProp / ChemBERTa: Academic ML toolkits for molecular property prediction, often used in research settings. These aren’t toxicology-specific but have been adapted to endpoints like hERG inhibition or mutagenicity.
The ML Evolution: From Classic Models to Deep Learning
Let's take a quick journey through the evolution of machine learning in toxicology.
First Wave: Classic ML Workhorses
Before the deep learning hype train arrived, toxicologists were already getting impressive results with these tried-and-true approaches:
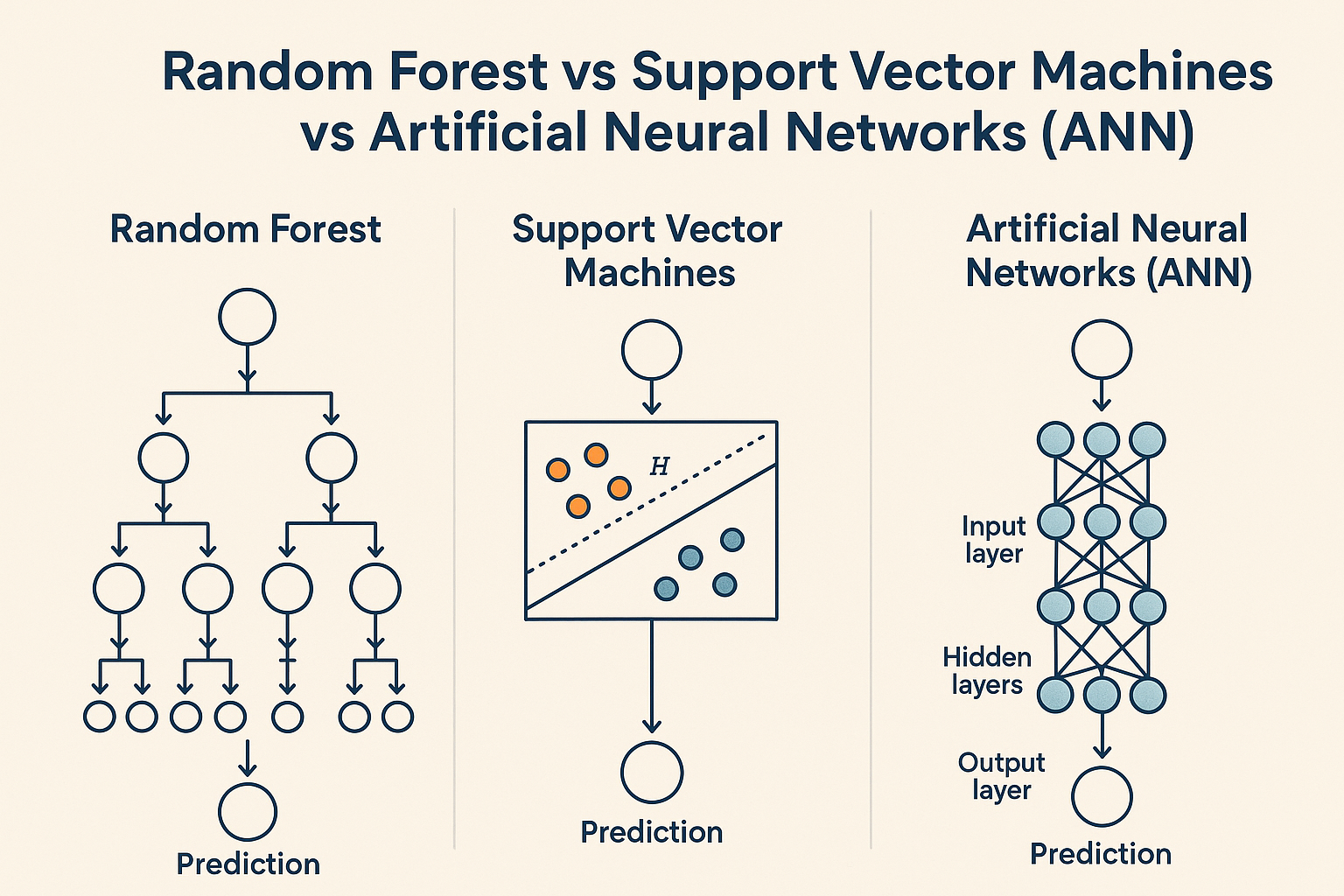
- Random Forests (RF): By combining multiple decision trees, they've proven remarkably effective for QSAR-type predictions while gracefully handling noisy datasets.
- Support Vector Machines (SVMs): Excel at binary classification (toxic vs. non-toxic). These were the go-to tools for early hERG cardiotoxicity screening, drawing clean boundaries between "safe" and "risky" compounds.
- Artificial Neural Networks (ANNs): The precursors to today's deep learning revolution. Even these simpler architectures showed impressive flexibility, though they demanded substantial training data.
What all these models had in common: they relied heavily on hand-crafted molecular descriptors, carefully calculated properties like logP (lipophilicity), polar surface area, or molecular fingerprints such as ECFP4 that capture structural patterns.
Second Wave: The Deep Learning Revolution
The game-changer? Modern deep learning architectures that bypass those human-designed descriptors entirely, learning directly from the raw molecular structure:
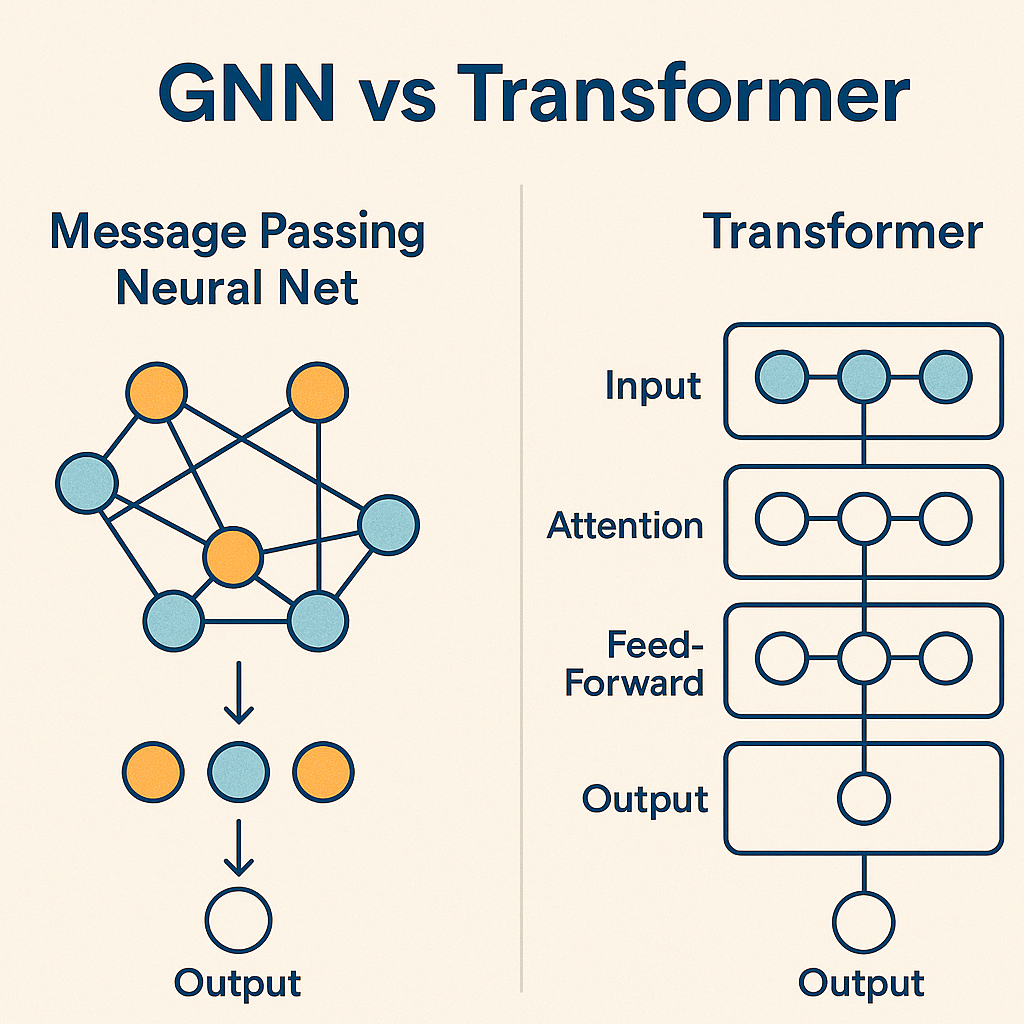
- Graph Neural Networks (GNNs): These treat molecules as they truly are graphs where atoms are nodes and bonds are edges. What makes GNNs revolutionary is their ability to discover complex structural patterns without being explicitly programmed. They can recognize dangerous motifs like "electron-rich aromatic ring adjacent to a nitro group" without a toxicologist spelling it out. They've become workhorses for predicting mutagenicity, cardiotoxicity, and liver damage. GNNs mimic how information flows through a molecule, with atoms "talking" to their neighbors through bonds. This captures the subtle electronic and structural dependencies that drive toxicity.
- Transformer-based Models: The same technology powering modern chatbots has been adapted for molecules. Models like ChemBERTa and MolBERT treat chemical structures (represented as SMILES strings) like sentences, learning the "grammar" of toxic compounds. These have achieved remarkable performance across the ADMET spectrum. Table 3: Strengths vs Weaknesses of ML/DL based methods
| Aspect | Strengths | Weaknesses |
| Interpretability | Advanced techniques like attention maps and SHAP values provide some insight | Often function as "black boxes" with limited explanation of predictions |
| Coverage | Can handle diverse chemical spaces and novel structural patterns | Heavily dependent on training data distribution and quality |
| Speed | Fast inference once trained; can process thousands of compounds quickly | Initial training can be computationally expensive and time-consuming |
| Validation | Often outperforms traditional methods in benchmark datasets | Regulatory acceptance still evolving; concerns about reproducibility |
| Integration | Modern APIs and containerization make deployment flexible | May require specialized infrastructure or expertise to implement |
| Data Requirements | Can extract patterns from complex, heterogeneous datasets | Performance degrades significantly with insufficient training examples |
| Adaptability | Can be retrained or fine-tuned as new data becomes available | May struggle with domain shift (e.g., novel chemical classes) |
The Full ADMET Gauntlet
Predicting off-target toxicity is crucial, but it’s only one piece of the survival game a drug has to play. To make it from bench to bedside, a molecule has to run the full ADMET gauntlet:
- Absorption – Can the drug actually get into the body (say, across the gut lining)?
- Distribution – Once inside, does it reach the right tissues at the right concentration?
- Metabolism – Does the liver shred it to pieces before it has a chance to work?
- Excretion – How efficiently does the body get rid of it, and through which routes?
- Toxicity – Does it cause collateral damage along the way?
You can have a perfectly targeted drug, but if it has poor solubility the project fails. This is why modern computational platforms predict the complete ADMET profile simultaneously, offering a comprehensive assessment of "drug-likeness." Multi-task learning models use a single molecular representation to predict multiple properties at once, rather than creating separate models for each characteristic.
This approach recognizes the interconnected nature of these properties:
- Lipophilicity influences both absorption and metabolic stability
- Plasma protein binding affects distribution and clearance
- Metabolic processes directly impact toxicity profiles
By training models to predict multiple properties simultaneously, we capture biological interdependencies that create more generalizable models representing how molecules behave as complete systems. This represents an evolution from isolated property analysis to understanding drugs as dynamic entities in biological systems.
Today's Toxicology Prediction
The real magic happens when these computational methods join forces in integrated screening workflows. Rather than betting on a single algorithm, modern pipelines create a consensus safety profile by combining multiple prediction strategies.
Take the Off-Target Safety Assessment (OTSA) framework - it's basically computational toxicology's version of a Swiss Army knife. It merges 2D similarity searches, QSAR models, and 3D pocket analysis into one automated system. Its secret weapon? It doesn't just analyze the drug itself but also its metabolites. Smart move, since those breakdown products often cause the actual toxicity. Here's a quick rundown of the tools toxicologists are using right now:
| Tool | Approach | Cost | What It Does Best |
| ADMET Predictor | AI/ML + QSAR | Commercial | All-in-one package with comprehensive risk scoring |
| Derek Nexus | Rule-Based | Commercial | Spots toxic fragments and explainswhythey're dangerous |
| ProTox-II | ML + Fragment-based | Free (Web) | Predicts organ-specific toxicity and mechanistic pathways |
| pkCSM | Graph-based ML | Free (Web) | Models complete ADMET profiles using molecular signatures |
Industry‑standard tools at a glance
- TEST (EPA Toxicity Estimation Software Tool):Ensemble QSAR. Combines multiple statistical models built on curated datasets and molecular descriptors, then uses a consensus prediction.Strengths: transparent descriptor-based rationale and EPA familiarity. Best for early hazard screening and benchmarking when you want quick, explainable calls.
- OECD QSAR Toolbox:Read‑across and chemical category formation. Uses profilers and mechanistic knowledge to group similar substances, infer properties, and document applicability domains and mechanistic hypotheses.Strengths: regulatory‑accepted workflows and rich provenance. Best for dossiers, justification of predictions, and mechanistic reasoning.
- LeadscopeKnowledge‑based structural alerts plus statistical modeling over large, curated toxicology corpora.Strengths: broad endpoint coverage and documentation supporting regulatory review. Best for hazard identification and safety assessment with traceable evidence.
When to use what (quick guide)
- Need explainable, regulator‑friendly rationale fast → Derek Nexus, TEST, Leadscope, OECD QSAR Toolbox.
- Need broad ADMET coverage across endpoints at once → ADMET Predictor, pkCSM, ProTox‑II.
- Need mechanistic or target‑specific insight → Docking or reverse docking, then cross‑check with rule‑based alerts and ML.
- Preparing a dossier or justification package → OECD QSAR Toolbox and Leadscope for provenance plus consensus with ML outputs.
Beyond Molecules: The Systems View
Finding that your compound binds to an off-target protein is just the beginning. The million-dollar question is: "Will this actually hurt someone?"
Enter systems toxicology - where we model how a molecular hiccup cascades through biological networks. This approach connects the dots between a single binding event and clinical adverse reactions by tracing the ripple effects through genes, proteins, and pathways.
Real-world example: You predict your drug binds to a certain protein. Great, but is that protein even expressed in liver tissue where toxicity might occur? Systems approaches layer in this crucial biological context.
Despite all this progress, some tough challenges remain:
- The Black Box: AI is powerful but often can't explain its predictions. Regulators (rightfully) demand to know why a model flags something as toxic.
- Garbage In, Garbage Out: Even the fanciest AI can't overcome flawed training data. The field desperately needs standardized, high-quality datasets.
- Regulatory Hurdles: Getting the FDA to accept computer predictions as primary evidence remains an uphill battle requiring rock-solid validation and transparency.
The future is clear: we're headed toward integrated platforms that combine in silico predictions with lab data in AI-powered frameworks that speak the language regulators understand.
Conclusion
Drug development is a high-stakes game where toxicity can sink even the most promising candidates. The computational tools we've discussed aren't just nice-to-have - they're essential for designing safer drugs more efficiently. We've evolved from the naive "magic bullet" idea to understanding that drugs interact with multiple targets throughout the body. Our computational arsenal now lets us predict these complex interaction profiles before spending a dime on synthesis.
While challenges in data quality and model interpretability persist, the trajectory is promising. By catching problems early through computational prediction, we're steering drug discovery toward molecules with better odds of success - ultimately delivering safer medicines to the patients who need them.
We are on a mission to make molecular and structural biology related experiments easier than ever. Whether you are doing research on protein design, drug design or want to run and organize your experiments, LiteFold helps to manage that with ease. Try out, it's free.