The Generative Geometric Turn in AI Drug Discovery


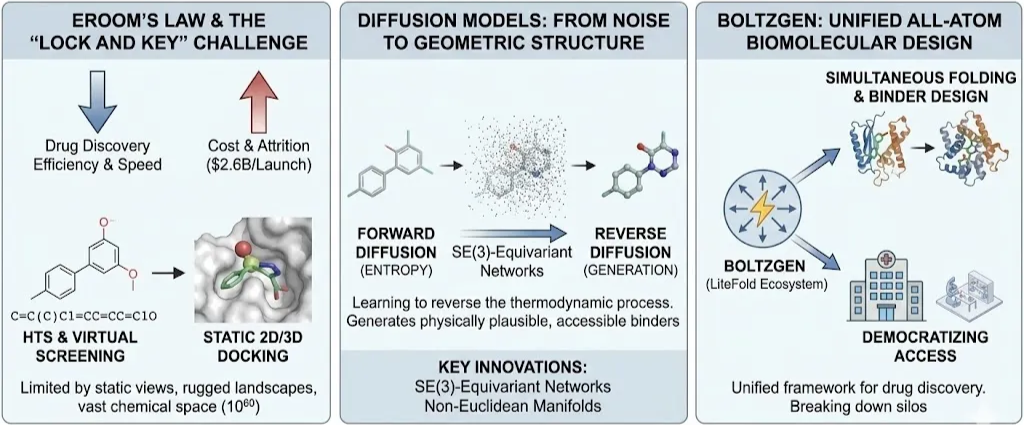
The pharmaceutical industry stands at a critical juncture, often described through the lens of "Eroom's Law"—the observation that drug discovery is becoming slower and exponentially more expensive over time, despite aggregate improvements in technology. The process of identifying a therapeutic candidate, optimizing its properties, and guiding it through clinical trials is historically a venture of immense attrition, costing upwards of $2.6 billion per successful launch. Central to this inefficiency is the "lock and key" problem: finding a small molecule (ligand) that binds with high affinity and specificity to a biological target (protein), typically by fitting into a complex, three-dimensional binding pocket. For decades, this challenge was addressed through High-Throughput Screening (HTS) of physical libraries or, more recently, virtual screening using classical docking algorithms like AutoDock Vina or Glide. While these methods have yielded successes, they are fundamentally limited by their reliance on static representations, rugged energy landscapes, and the sheer vastness of chemical space estimated at over 1060 pharmacologically active compounds.
A profound shift is now underway, driven by the integration of geometric deep learning and generative artificial intelligence. This "Geometric Turn" acknowledges that molecules are not merely strings of text (SMILES) or 2D graphs, but dynamic physical objects embedded in continuous Euclidean space. The most potent engine of this revolution is the Diffusion Model. Originally popularized in computer vision for generating photorealistic images from noise, diffusion models have been mathematically adapted to the non-Euclidean manifolds of molecular geometry. By learning to reverse the thermodynamic process of entropy turning structure into noise and back again these models are enabling the de novo generation of binders that are physically plausible, synthetically accessible, and tailored to novel targets that have historically been deemed "undruggable".
We are on a mission to make molecular and structural biology related experiments easier than ever. Whether you are doing research on protein design, drug design or want to run and organize your experiments, LiteFold helps to manage that with ease. Try out, it's free.
This Blog provides an exhaustive technical analysis of the application of diffusion models to Small-Molecule Generation and Binding Pose Prediction. We will dissect the theoretical underpinnings of SE(3)-equivariant networks, analyze the landscape of current state-of-the-art architectures (from DiffDock to TargetDiff), and finally, explore the next frontier in unified biomolecular design: the BoltzGen. As part of the LiteFold ecosystem, BoltzGen represents a paradigm shift from isolated task specific models to a unified all atom generative framework capable of simultaneous folding and binder design, democratizing access to the kind of computational infrastructure that was once the exclusive preserve of big pharma.
Theoretical Foundations: Thermodynamics and Geometric Deep Learning
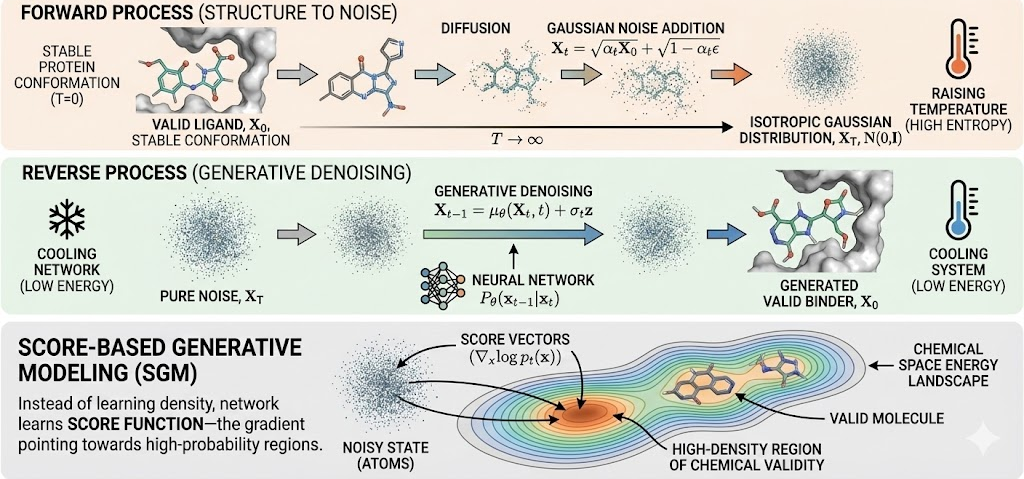
To understand the efficacy of diffusion models in chemistry, one must first grasp their statistical mechanical roots. Unlike Generative Adversarial Networks (GANs), which rely on unstable adversarial training, or Variational Autoencoders (VAEs), which frequently suffer from posterior collapse, diffusion models are grounded in a stable, iterative denoising process inspired by non-equilibrium thermodynamics. The framework operates through two dual processes:
1. The Forward Process (Diffusion): This is a fixed Markovian chain that progressively destroys structure. Given a data distribution X0 (e.g., a valid crystal structure of a ligand), Gaussian noise is added over discrete time steps t=0…..T. As t to T, the molecular geometry dissolves into an isotropic Gaussian distribution N(0,1). This process is chemically equivalent to raising the temperature of a system until it becomes a high-entropy gas.
2. The Reverse Process (Generative Denoising): The generative model learns to reverse this entropy. Starting from pure noise sampled from a standard Gaussian, a neural network approximates the reverse transition Pθ(xt-1|xt), effectively "cooling" the system to recover a low-energy, chemically valid state.
In the context of Structure-Based Drug Design (SBDD), this is often formulated as Score-Based Generative Modeling (SGM). Instead of trying to learn the intractable probability density function of all possible molecules, the network learns the "score function"—the gradient of the log-density of the data (∇xlog pt(x)). Intuitively, this vector field points the "atoms" from their noisy positions toward high-density regions of chemical validity.
The Non-Negotiable Requirement: SE(3) Equivariance
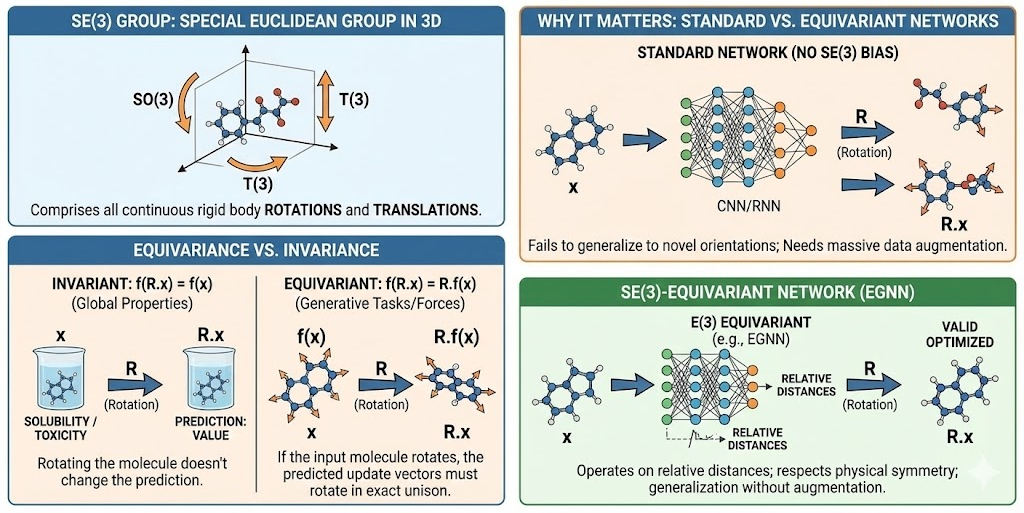
Applying neural networks to 3D chemistry introduces a constraint that does not exist in text or image generation: the laws of physics must be invariant to the observer's frame of reference. A molecule's internal energy, bond lengths, and binding affinity do not change if the molecule is rotated 90 degrees or translated 5 angstroms to the left.
Standard neural architectures (CNNs, RNNs) are not naturally invariant to 3D rotations. If a standard network is trained on a molecule, and that molecule is rotated, the network might perceive it as a completely different object. This necessitates SE(3) Equivariance
- SE(3) Group: The Special Euclidean group in 3 dimensions, comprising all continuous rigid body translations and rotations.
- Equivariance vs. Invariance:
- Invariant: f(R.x) = f(x) Global properties like "predicted toxicity" or "solubility" should be invariant; rotating the molecule shouldn't change the prediction.
- Equivariant: f(R.x) = R. f(x). Generative tasks that predict atomic coordinates or forces must be equivariant. If the input molecule rotates, the predicted update vectors must rotate in exact unison.
The first deployment of diffusion models for 3D molecular generation, the E(3) Equivariant Diffusion Model (EDM), utilized E(n)-equivariant Graph Neural Networks (EGNNs). These networks operate on relative distances rather than absolute coordinates, ensuring that the generative process respects the symmetry of physical space. Without this inductive bias, models require massive data augmentation (showing the network every possible rotation of a molecule) and still fail to generalize to novel orientations.
Diffusion on Non-Euclidean Manifolds
Molecules are constrained systems. Atoms are not free-floating points; they are tethered by covalent bonds with specific lengths and rigid angles. Modeling a molecule purely in Euclidean space (R3) often leads to "broken" geometry carbon rings that are not flat, or bonds that stretch physically impossible distances.
Advanced diffusion approaches, particularly in molecular docking (e.g., DiffDock), operate on the product manifold of the ligand's degrees of freedom:
M= R3 x SO(3) x Tm
Where:
- R3 represents global translation.
- SO(3) represents global rotation (Special Orthogonal group).
- Tm represents the $m$ rotatable torsion angles (modeled as a hypertorus).
By defining the diffusion process directly on this manifold, the model ensures that the local geometry (bond lengths/angles) remains rigid and chemically valid, while the system explores the conformational space (folding/twisting) and the pose space (position/orientation) stochastically.
The Landscape of Small-Molecule Generation
The application of diffusion models to small molecules is broadly divided into two domains: Unconditional Generation (exploring chemical space for novel scaffolds) and Conditional Generation (designing ligands to fit a specific protein pocket SBDD).
Unconditional Generation: From Clouds to Graphs
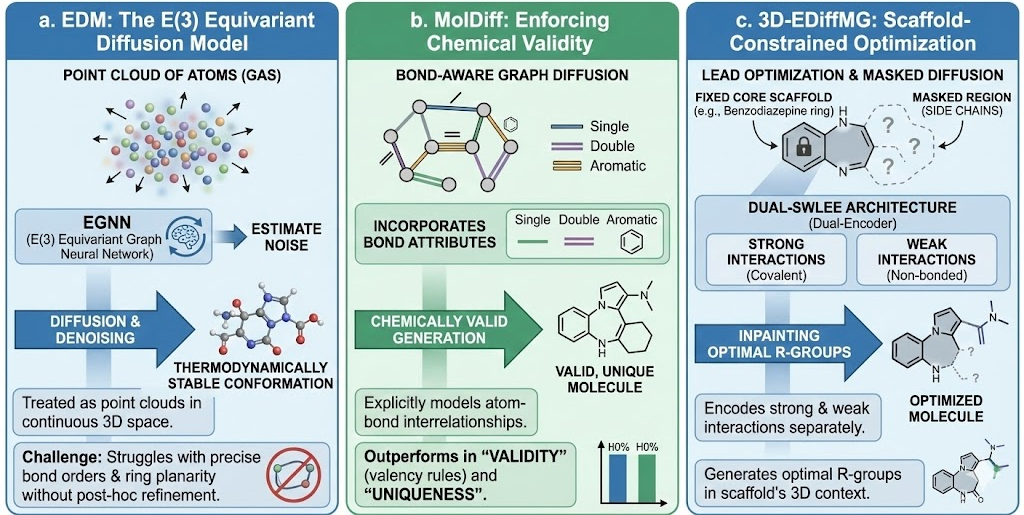
a. EDM: The E(3) Equivariant Diffusion Model
The E(3) Equivariant Diffusion Model (EDM) was a foundational architecture that treated molecules as point clouds of atoms in continuous 3D space. Using an EGNN to estimate noise, EDM demonstrated that diffusion models could generate thermodynamically stable conformations better than previous VAE or Flow based methods. However, because it treated atoms essentially as a "gas" that condenses into a molecule, it sometimes struggled with precise integer bond orders and ring planarity without post-hoc refinement.
b. MolDiff: Enforcing Chemical Validity
To address the limitations of pure point-cloud generation, MolDiff introduced a bond-aware diffusion framework. MolDiff explicitly models the interrelationships between atoms and bonds during the generative process. By incorporating bond attributes (single, double, aromatic) into the graph diffusion, it constrains the generation to chemically valid graphs. This approach significantly outperforms EDM in terms of "validity" (percentage of generated structures that respect valency rules) and "uniqueness," ensuring the model isn't just memorizing the training set.
c. 3D-EDiffMG: Scaffold-Constrained Optimization
In practical drug discovery, chemists rarely start from scratch. They often engage in Lead Optimization, where a core scaffold (e.g., a benzodiazepine ring) is known to bind, and the goal is to modify side chains to improve solubility or reduce toxicity. 3D-EDiffMG utilizes a dual-encoder architecture (Dual-SWLEE) that can encode strong interactions (covalent bonds) and weak interactions (non-bonded forces) separately. This allows for masked diffusion or "inpainting," where the core scaffold is fixed, and the diffusion model generates optimal R-groups in the context of the scaffold's 3D geometry.
Structure-Based Drug Design (SBDD)
The "Holy Grail" of generative chemistry is SBDD: given a protein pocket P, generate a ligand L that maximizes the probability p(L|P).
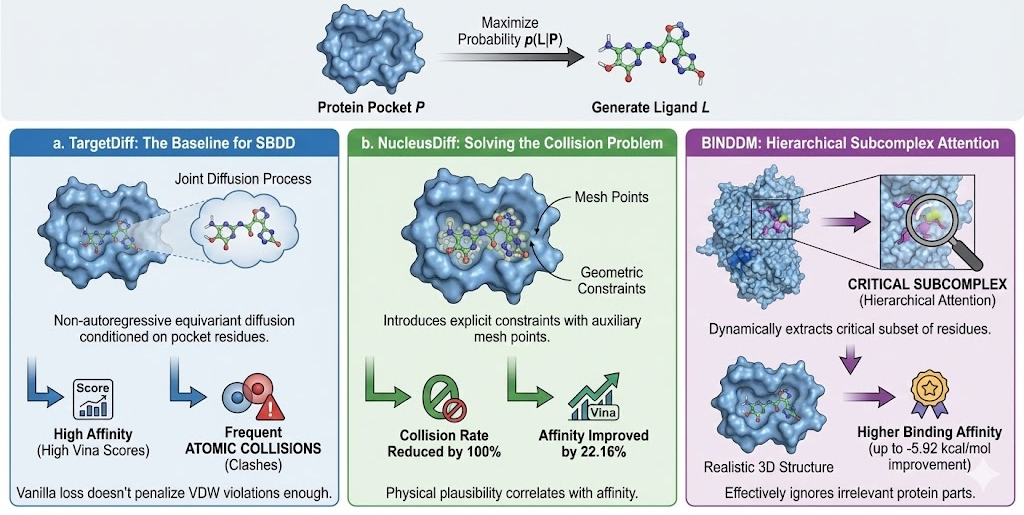
a. TargetDiff: The Baseline for SBDD
TargetDiff represents the first successful application of equivariant diffusion to non-autoregressive SBDD. It defines a joint diffusion process over atom types and coordinates, conditioned on the protein pocket's residues. By modeling the interaction as a distribution, TargetDiff captures the multi-modal nature of binding—there isn't just one molecule that fits a pocket, but a diverse family of potential binders. However, early evaluations showed that while TargetDiff generated high-affinity ligands (high Vina scores), it often produced molecules with atomic collisions (clashes) with the protein, as the vanilla diffusion loss didn't penalize van der Waals violations heavily enough.
b. NucleusDiff: Solving the Collision Problem
Atomic collisions render a docked pose physically impossible (infinite energy). NucleusDiff addresses this by introducing explicit geometric constraints into the diffusion process. It places auxiliary "mesh points" on the spherical van der Waals surface of each atom. The loss function includes a regularization term that enforces minimum distances between these mesh points and the protein atoms.
- Result: NucleusDiff reduces the atomic collision rate by up to 100% compared to TargetDiff.
- Affinity: It simultaneously improves the average Vina score by 22.16% on the CrossDocked2020 benchmark, proving that physical plausibility correlates with predicted affinity.
BINDDM: Hierarchical Subcomplex Attention
Proteins are large macromolecules; generating a small ligand while attending to thousands of protein atoms creates signal-to-noise issues. BINDDM (Binding-Adaptive Diffusion Model) employs a hierarchical approach. It dynamically extracts a "subcomplex" the subset of residues most critical for binding and focuses the SE(3)-equivariant network's attention there. This "zoom-in" mechanism allows BINDDM to generate ligands with more realistic 3D structures and higher binding affinities (up to -5.92 kcal/mol improvement in Vina score) by effectively ignoring the irrelevant parts of the protein surface.
The Revolution in Molecular Docking: From Search to Generation
While SBDD focuses on generating new molecules, Molecular Docking focuses on predicting the binding pose of existing molecules. This is the workhorse of virtual screening.
a. The Limitations of Traditional Algorithms
Traditional docking tools like AutoDock Vina, Glide, and Surflex-Dock treat docking as an optimization problem on a rugged energy landscape.
- Mechanism: They use a scoring function (physics-based or empirical) and a search algorithm (Genetic Algorithm, Monte Carlo, or Simulated Annealing) to find the global minimum.
- Failures: These search algorithms frequently get trapped in local minima. They are computationally expensive (minutes per ligand), scaling poorly to billion-compound libraries. Furthermore, they perform poorly on "blind docking" (where the pocket location is unknown) because the search space is too vast.
b. DiffDock: A Generative Paradigm Shift
DiffDock (Diffusion Docking) reframes the problem entirely. It does not "search" for a minimum energy; it "generates" the binding pose distribution.
- Architecture: DiffDock operates on the product manifold of translations, rotations, and torsions.
- Process: It samples random poses (noise) and iteratively refines them using a learned score model that "pushes" the ligand toward the pocket.
- Confidence Model: Crucially, DiffDock includes a confidence model that ranks the generated poses. Since diffusion is stochastic, it can generate multiple hypotheses; the confidence model predicts which one is correct.
Benchmarking Dominance: On the PDBBind benchmark, DiffDock achieves a 38% Top-1 success rate (RMSD < 2Å), significantly outperforming traditional docking (23%) and previous deep learning regression models like EquiBind (20%). Even more impressively, its inference time is orders of magnitude faster than exhaustive search methods, making it feasible for library-scale screening.
c. DiffDock-PP and Protein-Protein Interactions
The principles of DiffDock have been extended to DiffDock-PP for rigid protein-protein docking. Protein-protein interactions (PPIs) are notoriously difficult due to the large surface areas and subtle energetics. DiffDock-PP frames PPI docking as learning a probability distribution over the SE(3) transformation that aligns the ligand protein to the receptor. It runs 5 to 60 times faster than traditional methods on GPU hardware, opening the door for interactome-scale mapping.
Challenges and Frontiers
Synthetic Accessibility (SA)
A recurring criticism of generative AI in chemistry is the "hallucination" of unsynthesizable molecules. A model might propose a structure that binds perfectly in silico maximizing Vina scores but contains highly strained rings, impossible bond angles, or functional groups that would be chemically unstable or impossible to synthesize in a lab.
- The Metric: Synthetic Accessibility (SA) score is a heuristic ranging from 1 (easy) to 10 (hard), penalizing complexity, chiral centers, and fused rings.
- Integration: Modern pipelines like LiteFold integrate SA scoring directly into the generation loop. By using classifier guidance or filtering, the diffusion process can be steered away from high-SA regions of chemical space, ensuring that the "fail fast" philosophy applies to synthesis planning as well as binding.
The "Static Receptor" Fallacy vs. Co-Folding
Most current docking and SBDD models assume a "rigid receptor" the protein is treated as a static statue. In biological reality, proteins are breathing, dynamic entities. Binding often involves induced fit (the pocket reshapes to accommodate the ligand) or conformational selection.
- The Problem: Rigid docking fails when the holo (bound) structure of the protein differs significantly from the apo (unbound) structure.
- The Solution: The field is moving toward Co-Folding models (like AlphaFold Multimer and Boltz-1), which predict the structure of the protein and ligand simultaneously from sequence. This allows the model to predict the induced fit changes, offering a higher fidelity representation of the interaction.
The LiteFold Ecosystem and the BoltzGen Pipeline
The fragmentation of the current AI drug discovery landscape where a researcher needs one tool for folding, another for docking, and a third for design creates immense friction. LiteFold aims to unify this stack, positioning itself as the "Infrastructure for Drug Discovery." By providing high-performance compute, secure data handling, and browser-based interfaces for complex MD and docking simulations, LiteFold democratizes access to these advanced capabilities.
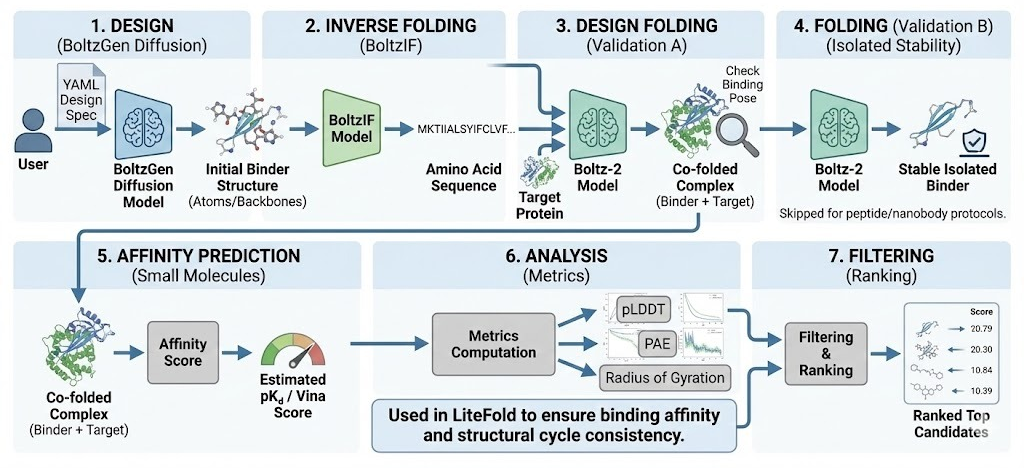
The crown jewel of this ecosystem is the upcoming BoltzGen pipeline, a unified all-atom generative framework that integrates the state of the art Boltz-1 biomolecular interaction model.
BoltzGen: Unifying Design and Structure Prediction
BoltzGen is not merely a docking tool; it is a generative engine that unifies design and structure prediction. Developed in collaboration with MIT’s Jameel Clinic and validated by major biotech partners, it represents a departure from the "pipeline of distinct models" approach.
Architectural Innovation: Geometry-Based Residues
Unlike language models that treat amino acids as text tokens, BoltzGen employs a purely geometry-based representation of designed residue types.
- The model predicts the geometric cluster of atoms that constitutes a residue (e.g., the specific cloud of atoms that makes an Alanine).
- This allows the model to train on structure prediction (folding) and design (generation) simultaneously. The gradient flow is continuous, enabling the model to "reason" about steric clashes and packing density in real time during generation.
- This unification allows BoltzGen to match the folding performance of state of the art predictive models (like Boltz-1) while retaining the creativity of a generative model.
Unprecedented Performance on Novel Targets
The true test of any AI model in biology is generalization. Models often "memorize" the PDB, performing well on familiar targets but failing on novel ones.
- The Benchmark: BoltzGen was tested on 9 novel targets that have <30% sequence identity to any complex in the PDB (effectively "aliens" to the model).
- The Result: BoltzGen successfully designed nanomolar (nM) binders for 66% of these targets (6 out of 9).
- Efficiency: This was achieved by testing fewer than 15 designs per target in the wet lab.
This efficiency, a hit rate of >5% on the very first batch of <15 designs is orders of magnitude better than traditional HTS (hit rates often <0.1% of thousands of compounds) and earlier AI generation methods. It suggests that BoltzGen has learned a generalized "physics of binding" rather than just memorizing specific interaction motifs.
Multi-Modal Capabilities
BoltzGen is modality-agnostic. Through its Design Specification Language, users on LiteFold can define constraints for:
- Proteins & Nanobodies: Designing large biologic binders for immunotherapy.
- Peptides: Designing cyclic peptides or macrocycles for "undruggable" flat interfaces (e.g., PPIs).
- Small Molecules: While primarily a protein design model, the pipeline supports small molecule contexts, allowing for the design of protein binders to small molecules (e.g., biosensors) or vice-versa.
Operational Excellence: LMI4Boltz and Hardware Optimization
Deploying these massive all-atom models requires significant computational resources. The standard Boltz-2 model is VRAM-hungry, limiting inference on consumer hardware. The LiteFold implementation leverages LMI4Boltz (Low Memory Inference for Boltz), a set of optimizations including:
- Tensor Offloading: Moving large, infrequently used tensors (like MSA representations) to host memory.
- In-Place Operations: Using FlashAttention and in-place updates to avoid memory spikes.
- Result: These optimizations increase the token limit by 66.7%, allowing for the modeling of large complexes (>2,660 tokens) on standard 24GB VRAM GPUs (like the A10G or 3090/4090), making the pipeline accessible to a broader range of researchers via the LiteFold cloud.
Comparative Benchmarking: Boltz-1 vs. AlphaFold3
As the foundation of BoltzGen, the Boltz-1 model's accuracy is paramount. In comparative benchmarks using ABCFold standards:
- Accuracy: Boltz-1 achieves AlphaFold3-level accuracy in predicting 3D structures of biomolecular complexes.
- CASP15 Performance: On the critical LDDT-PLI metric (protein-ligand interactions), Boltz-1 achieves a score of 65%, matching AlphaFold3 and significantly outperforming Chai-1 (40%).
- Open Source: Unlike AlphaFold3, which is restricted, Boltz-1 is fully open-source (MIT License), allowing LiteFold to integrate it deeply into the generation pipeline without API restrictions or data privacy concerns.
| Model | Core Mechanism | Input Data | Output | Key Innovation |
|---|---|---|---|---|
| EDM | E(3)-Equivariant Diffusion | Point Cloud | Atom Coordinates | First stable 3D molecular diffusion |
| MolDiff | Bond-Aware Diffusion | Graph + 3D | Graph + Coordinates | Enforces chemical validity & bond orders |
| TargetDiff | Conditional SE(3) Diffusion | Protein Pocket + Ligand | Ligand Structure | Jointly models atom types & coords in pocket |
| NucleusDiff | Constrained Diffusion | Protein Pocket + Ligand | Ligand Structure | Mesh-based collision constraints (<1% clashes) |
| DiffDock | Manifold Diffusion | Protein (Blind) + Ligand | Binding Pose | Diffusion on R3 x SO(3) x Tm |
| BoltzGen | Unified All-Atom Diffusion | Target + Constraints | Binder (Protein/Mol) | Simultaneous Design & Folding |
Conclusion: The Fail-Fast Future
The integration of BoltzGen into the LiteFold platform signals the maturation of generative biology. We are moving away from the era of "computer-aided drug design" (where computers acted as assistants to human intuition) to "computer-driven drug design" (where algorithms act as the primary architects).
By combining the stochastic exploration of Diffusion Models, the rigorous physical constraints of SE(3) Equivariance, and the validation power of Co-Folding within a unified pipeline, we can now "fail fast and fail cheap" in silico. Instead of synthesizing 10,000 compounds to find one hit, researchers can generate 100 high-confidence designs, validate them computationally with Boltz-2, and proceed to the wet lab with a focused set of 15 candidates that have a >60% probability of success.
This is not just an incremental improvement in efficiency; it is a fundamental restructuring of the economics of drug discovery. For the LiteFold community, the release of the BoltzGen pipeline is the key to unlocking this potential, transforming the browser into a portal for de novo biological engineering.
We are on a mission to make molecular and structural biology related experiments easier than ever. Whether you are doing research on protein design, drug design or want to run and organize your experiments, LiteFold helps to manage that with ease. Try out, it's free.