Structure Based Drug Design just got easier than ever



We present LiteFold DeNovo, our second flagship feature, a highly efficient structure based drug design workflow to accelerate your lead candidate research to the next level. In this blog, we will discuss the features that are available today, how to get started, and the new features that are coming very soon.
What is Structure Based Drug Design
If you are fairly new to this field and want to learn more about the basics of Structure Based Drug Design (SBDD), this section is for you.
For decades, drug discovery was akin to searching for a needle in a haystack only without knowing what the needle looked like or even if there was one to find. Scientists would test thousands of compounds against disease targets, hoping something would work, often with little understanding of why successful drugs succeeded or failed drugs didn't. This trial-and-error approach was expensive, time-consuming, and frequently led to dead ends after years of research.
Structure Based Drug Design (SBDD) transformed this process by turning on the lights. To put it simply, SBDD is the process of creating new molecules using the three dimensional structure of the binding pocket of the protein target. The binding pocket can be thought of as a lock that a molecule can fit snugly into. By using the three dimensional structure of the interior of the lock, molecules can be crafted to specifically match that complementary shape to perfectly fit that lock. The goal is to always maximally fit a particular lock while minimizing the amount of other locks that molecule can fit into – fitting into multiple binding pockets is a common cause of off-target effects and can lead to unintended side effects.
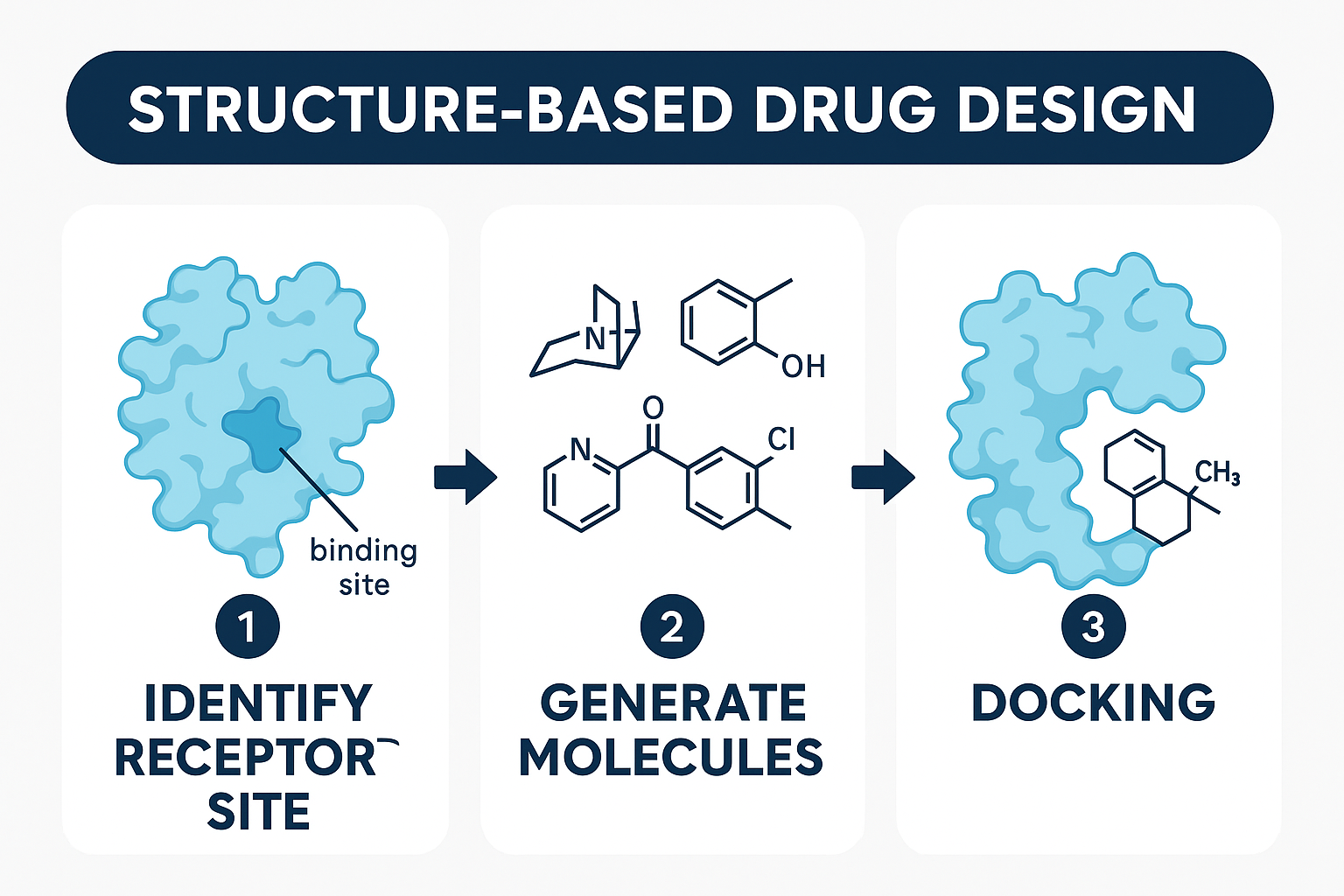
Why Structure Based Drug Design
This structure-guided approach offers dramatic advantages over traditional methods. Rather than synthesizing and testing thousands of random compounds, a process which can take years and cost millions, SBDD allows researchers to focus on molecules most likely to succeed. Traditional high-throughput screening might test a billion compounds to find a few promising leads, while SBDD can identify strong candidates from much smaller, rationally designed libraries. This means fewer failed experiments, reduced costs, and faster paths to potential therapies.
The power of SBDD has been proven repeatedly in breakthrough medications. HIV protease inhibitors like saquinavir, one of the first major SBDD successes, were designed by analyzing the precise structure of the HIV enzyme. Cancer drugs like imatinib (Gleevec) were crafted to fit perfectly into specific kinase binding sites. More recently, COVID-19 antivirals like nirmatrelvir (Paxlovid) were developed using SBDD principles, demonstrating how structural knowledge can accelerate drug development even under urgent timelines.
Beyond initial discovery, structural knowledge enables systematic optimization that's impossible with traditional approaches. When researchers see exactly how a molecule sits in its target binding site–which atoms interact where, which parts contribute to binding–they can methodically modify the drug to enhance potency, improve selectivity, or reduce side effects. It's like being able to file down specific parts of the key while watching exactly how it fits into the lock, rather than guessing which modifications might work. This rational optimization process can transform a weak initial hit into a potent, selective drug through cycles of structural analysis and informed design.
The success of SBDD raises an exciting possibility: if structural knowledge is so powerful, what if we could rapidly generate not just a few optimized molecules, but hundreds? LiteFold makes this possible by using neural network models to predict molecular structures specifically designed to fit into target pockets—completing predictions in minutes rather than hours or days. This rapid structure-based drug design approach allows our platform to generate hundreds of molecule candidates in a single workflow, enabling rational design of molecules with higher specificity, higher potency, and fewer off-target effects.
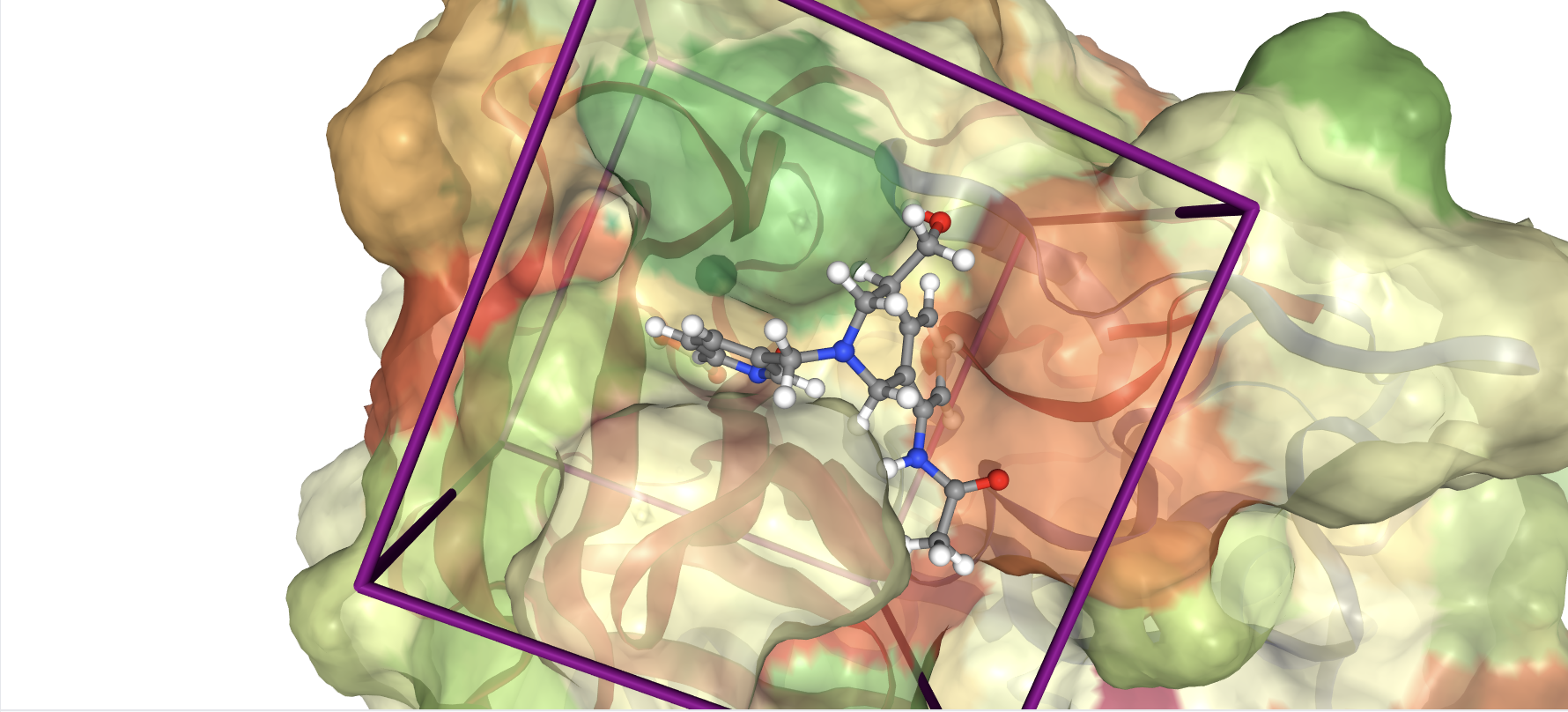
Example of structure-based molecular design: A computationally generated molecule (center) sits precisely within its target protein binding pocket (highlighted in purple box), demonstrating how structural knowledge enables the rational design of drug candidates with optimal fit and specificity.
We are on a mission to make molecular and structural biology related experiments easier than ever. Whether you are doing research on protein design, drug design or want to run and organize your experiments, LiteFold helps to manage that with ease. Try out, it's free.
How to use LiteFold DeNovo in your drug design workflow
To get started with LiteFold DeNovo, navigate to the DeNovo section inside the Lab tab.
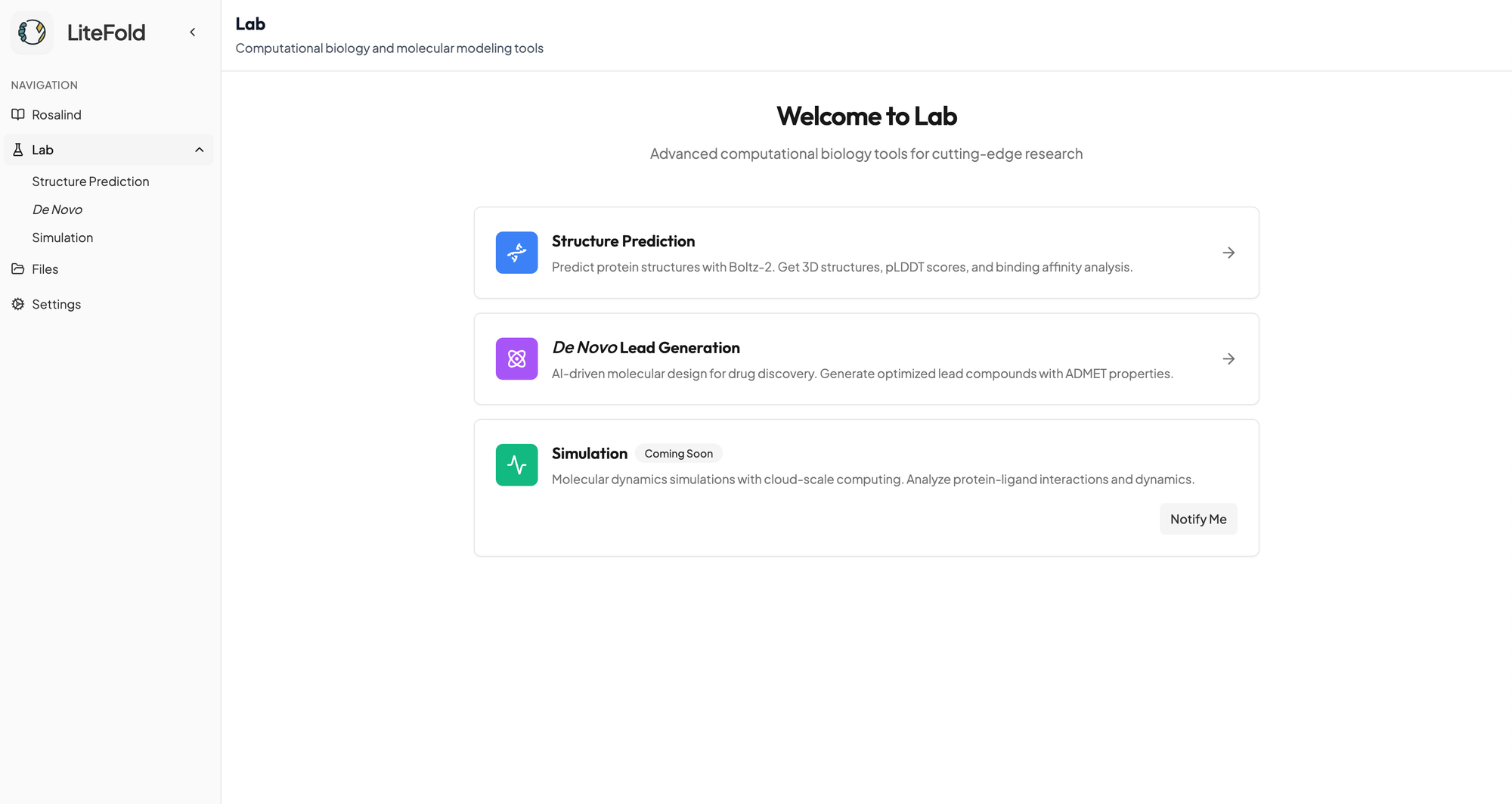
You can upload a PDB file of your own or use a protein structure previously predicted through our Structure Prediction pipeline. The platform will first guide you to choose a target pocket, which can be entered manually or our AI pocket prediction service will find and detect pockets that could be targeted for you.
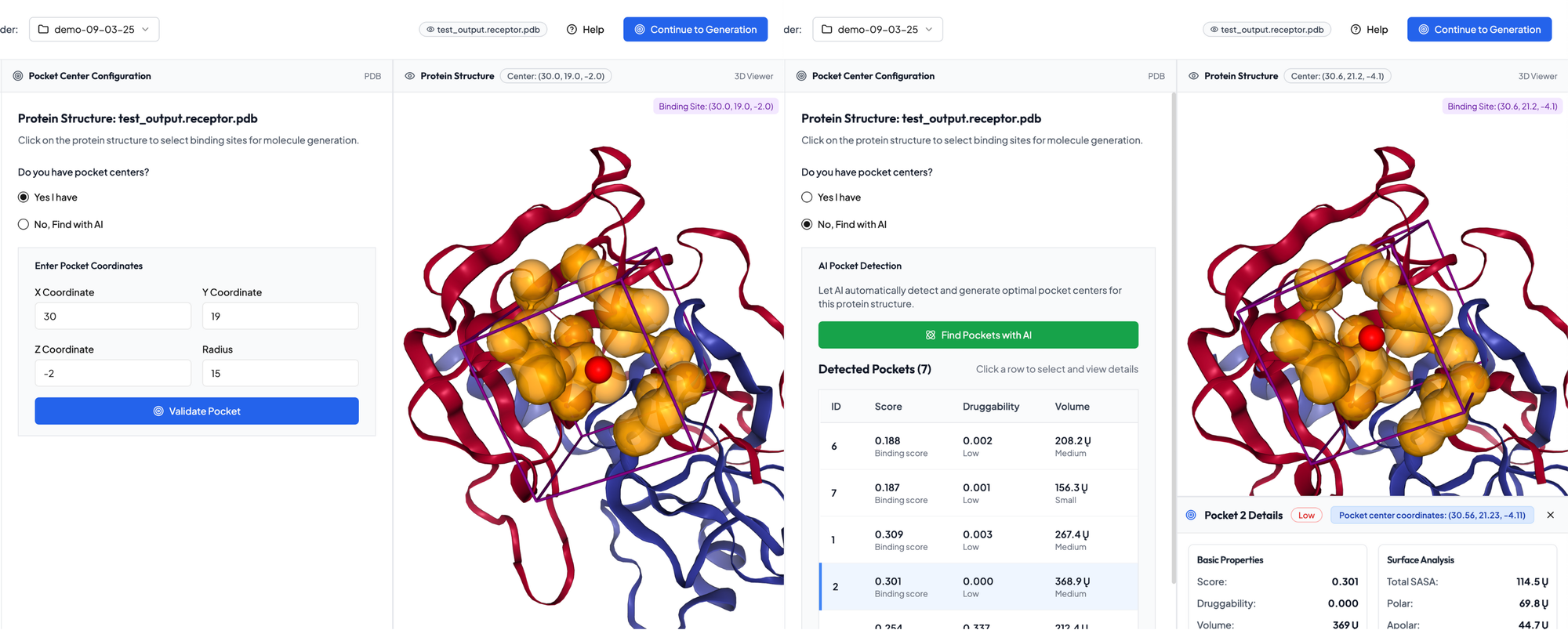
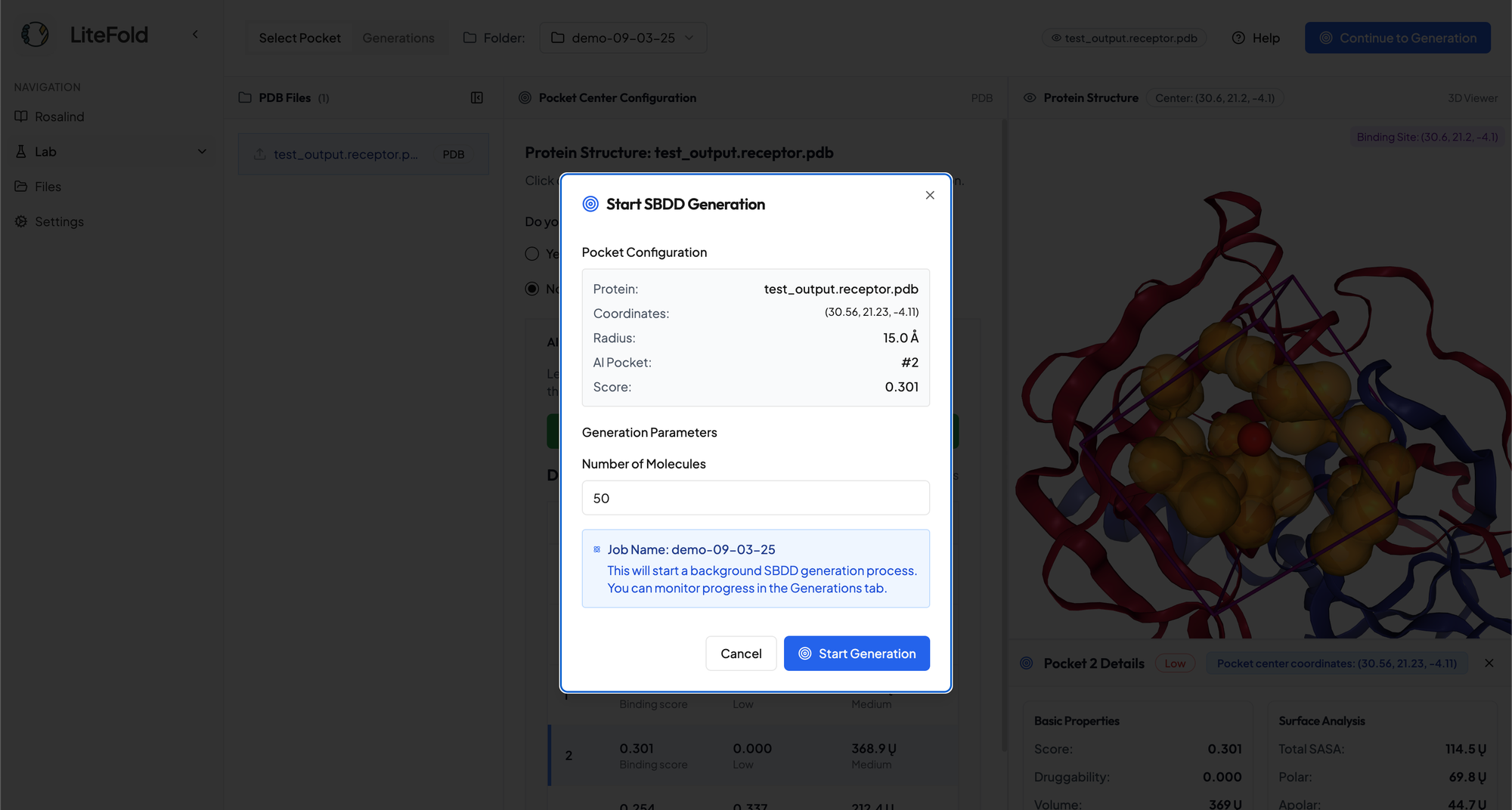
Now you are ready to generate molecules for your target pocket! While there is no limit to how many molecules you can generate, our free tier has a cap of 100 molecules.
Once the molecules are generated, you are presented with an intuitive, integrated workspace that transforms complex drug discovery into an accessible, visual experience. View your candidates in an interactive 3D window to see exactly how each molecule fits within the target binding pocket, then seamlessly switch to the built-in molecule editor to refine promising structures with just a few clicks. All critical metrics are displayed at a glance, allowing you to instantly identify the most promising candidates. This streamlined workflow accelerates your path from initial concept to lead compounds, condensing what traditionally takes weeks of computational work into a single, efficient session. With features like molecular dynamics simulation (coming soon!), you can validate and optimize your designs without switching between multiple software tools or waiting for external analysis.
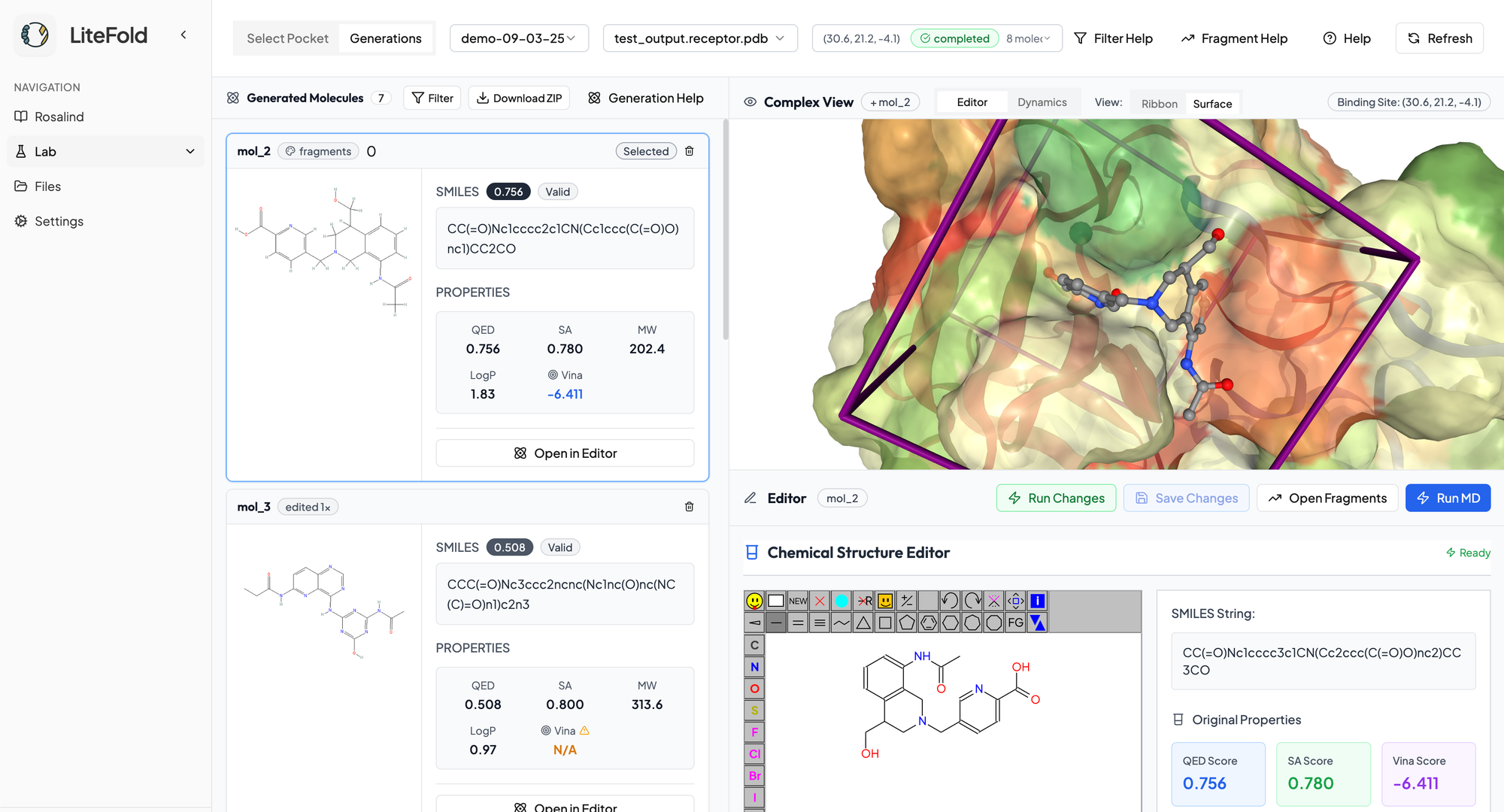
Fragment Growing & Molecule Editing
LiteFold DeNovo offers two ways to quickly edit the generated molecules: fragment growing and the molecule editor. Fragment growing allows you to take an already existing molecule and grow or add new pieces within the context of the binding pocket. The molecule editor allows you to make fine tuned adjustments by hand to change the structure of the molecule and then quickly visualize the changes. Any new changes to a molecule recomputes its metrics and binding affinity score so you can see if your edits increase the potency and drug-likeliness properties.
Supported Metrics
Each molecule generated has a few properties computed automatically: QED, Synthetic Accessibility, and Vina docking scores. The QED (Quantitative Estimate of Drug-likeness) score ranges from 0 to 1, with higher values indicating that a molecule possesses favorable drug-like properties such as appropriate molecular weight, lipophilicity, and other physicochemical characteristics that correlate with successful oral drugs.
The Synthetic Accessibility (SA) score predicts how challenging it would be to synthesize the molecule in the laboratory, with lower scores indicating easier synthetic routes—a crucial factor for determining whether promising candidates can be practically manufactured.
Finally, the Vina docking scores estimate the binding affinity between each generated molecule and the target protein, with more negative values suggesting stronger binding interactions and potentially higher potency. Together, these three metrics provide an immediate assessment of each candidate's drug-likeness, synthetic feasibility, and predicted efficacy, allowing researchers to quickly prioritize the most promising molecules for further development.
What's Next
In our next release, LiteFold will unveil a new and improved Rosalind and offer the ability for users to run molecular dynamic simulations!
The Simulation module will open the door for users to run different variations of molecular dynamics, spanning anything between long duration 1µs simulations and short 10ns stability measuring simulations (accelerated with metadynamics). This means you can actually watch how your molecules behave and how the protein reacts dynamically.
Rosalind's new upgrades will allow her to operate as a medicinal chemist. This includes editing molecules using her built-in chemical intuition, accessing the tools provided through LiteFold's Lab, and envisioning new mechanistic hypotheses for how compounds interact with their targets. She can iterate on designs, suggest improvements, and help you explore chemical space more efficiently than ever before.
Lastly, we are working on a High Throughput Virtual Screening workflow with Structure Based Virtual Screening (SBVS) and Ligand Based Virtual Screening (LBVS) options. This will bridge our technology stack with current state of the art extensive library filtering pipelines and improve upon them with user directed custom scoring functions; including pharmacophore, shape & electrostatic, and distance based similarity scores.
We can't wait to see the life-changing medicines you'll discover using these tools!
You can run some quick SBDD experiments at LiteFold, it's free. Let us know about your experience.